

Comparing the DNA hypermethylome with gene mutations in human colorectal cancer
- PMID: 17892325
- PMCID: PMC1988850
- DOI: 10.1371/journal.pgen.0030157
Comparing the DNA hypermethylome with gene mutations in human colorectal cancer
Abstract
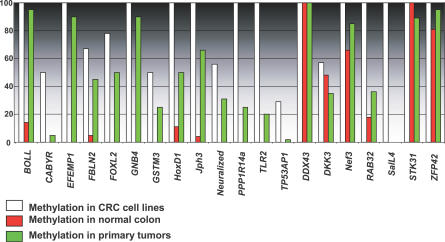
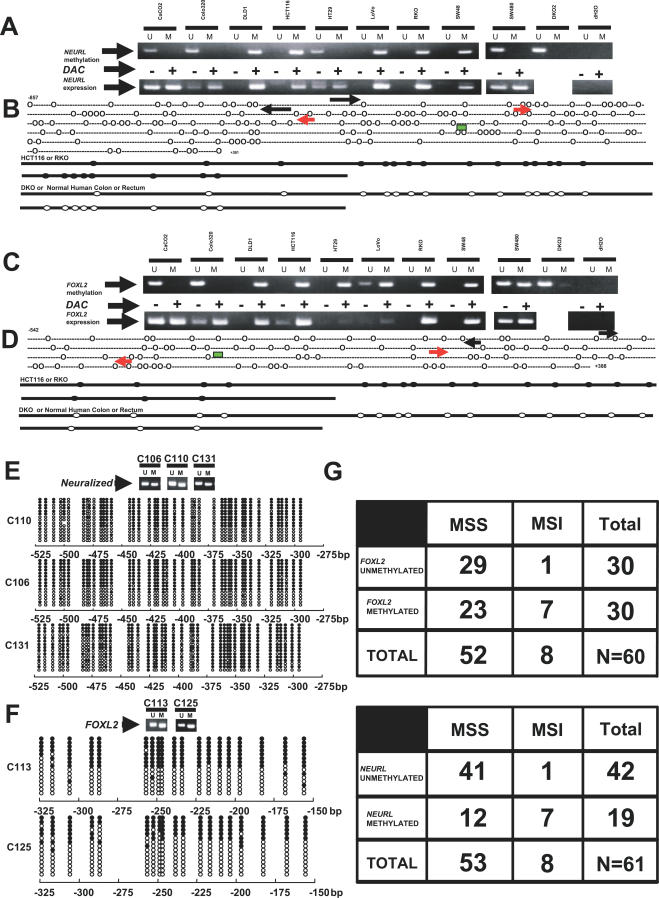
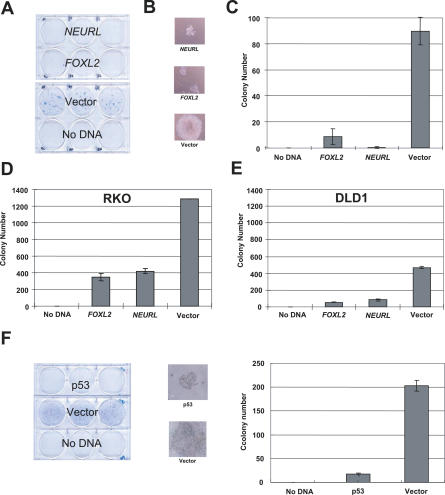
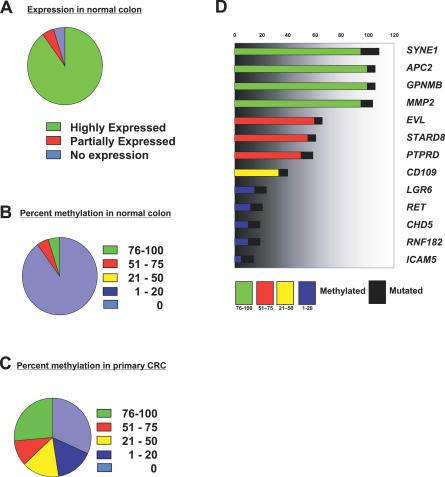
We have developed a transcriptome-wide approach to identify genes affected by promoter CpG island DNA hypermethylation and transcriptional silencing in colorectal cancer. By screening cell lines and validating tumor-specific hypermethylation in a panel of primary human colorectal cancer samples, we estimate that nearly 5% or more of all known genes may be promoter methylated in an individual tumor. When directly compared to gene mutations, we find larger numbers of genes hypermethylated in individual tumors, and a higher frequency of hypermethylation within individual genes harboring either genetic or epigenetic changes. Thus, to enumerate the full spectrum of alterations in the human cancer genome, and to facilitate the most efficacious grouping of tumors to identify cancer biomarkers and tailor therapeutic approaches, both genetic and epigenetic screens should be undertaken.
Conflict of interest statement
Competing interests. The commercial rights to the MSP technique belong to Oncomethylome Sciences. SBB and JGH serve as consultants to Oncomethylome Sciences and are entitled to royalties from any commercial use of this procedure.
Figures
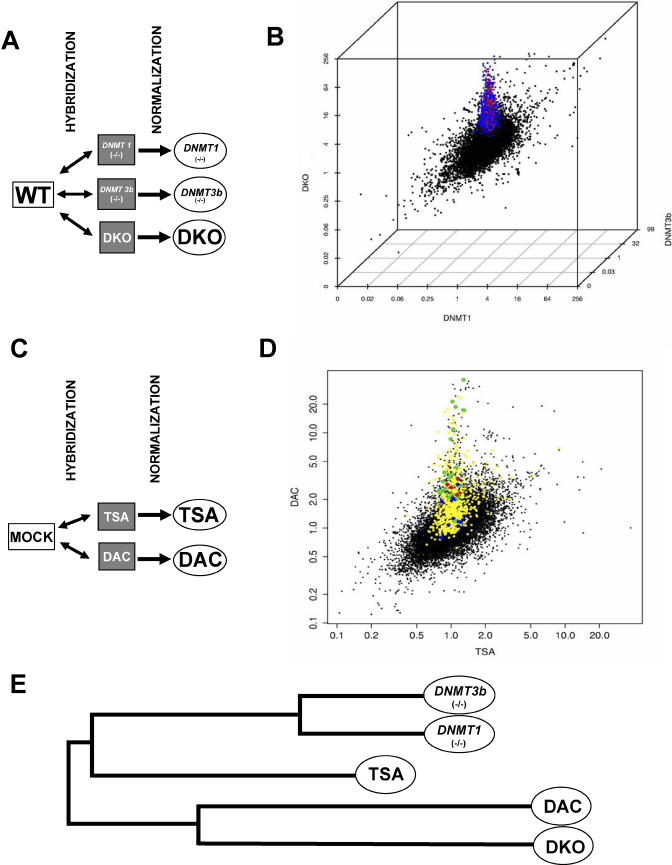
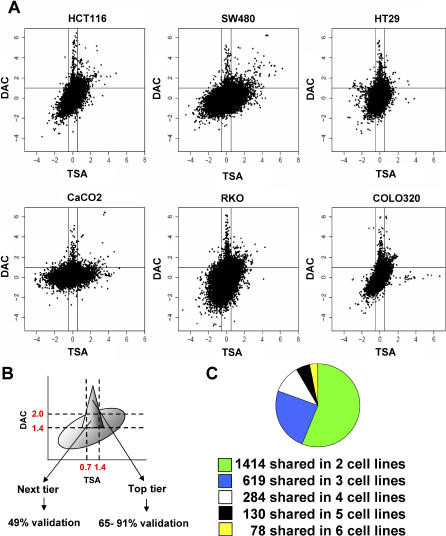
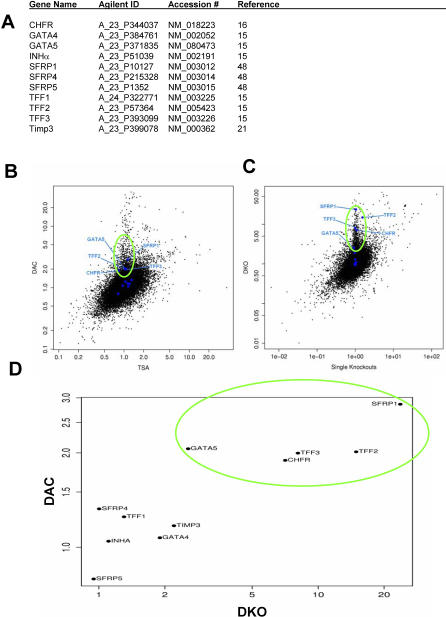

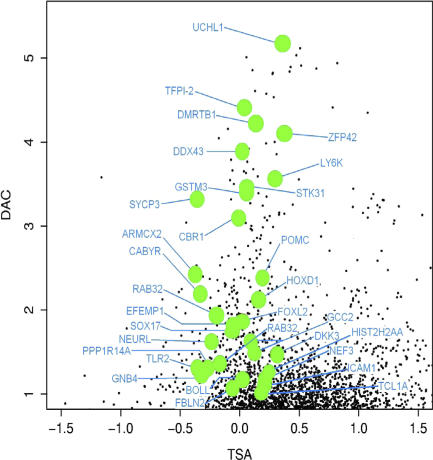


References
-
- Ponder BA. Cancer genetics. Nature. 2001;411:336–341. - PubMed
-
- Herman JG, Baylin SB. Gene silencing in cancer in association with promoter hypermethylation. N Engl J Med. 2003;349:2042–2054. - PubMed
-
- Sjoblom T, Jones S, Wood LD, Parsons DW, Lin J, et al. The consensus coding sequences of human breast and colorectal cancers. Science. 2006;314:268–274. - PubMed
-
- Costello JF, Fruhwald MC, Smiraglia DJ, Rush LJ, Robertson GP, et al. Aberrant CpG-island methylation has non-random and tumour-type-specific patterns. NatGenet. 2000;24:132–138. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
