

Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells
- PMID: 17894886
- PMCID: PMC2048942
- DOI: 10.1186/1471-2164-8-340
Comparison of the contributions of the nuclear and cytoplasmic compartments to global gene expression in human cells
Abstract
Background: In the most general sense, studies involving global analysis of gene expression aim to provide a comprehensive catalog of the components involved in the production of recognizable cellular phenotypes. These studies are often limited by the available technologies. One technology, based on microarrays, categorizes gene expression in terms of the abundance of RNA transcripts, and typically employs RNA prepared from whole cells, where cytoplasmic RNA predominates.
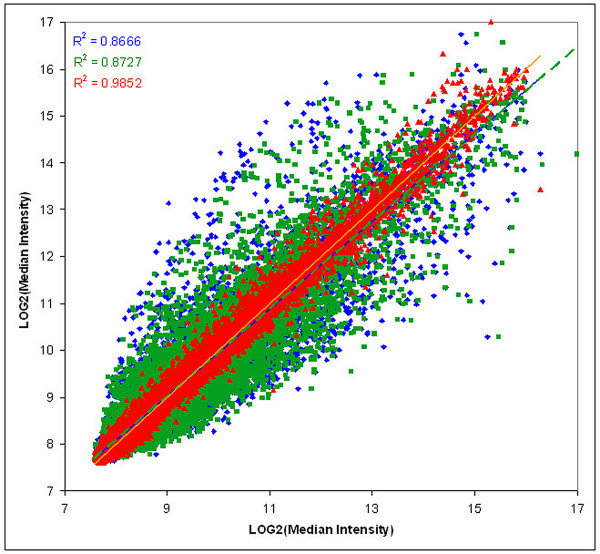
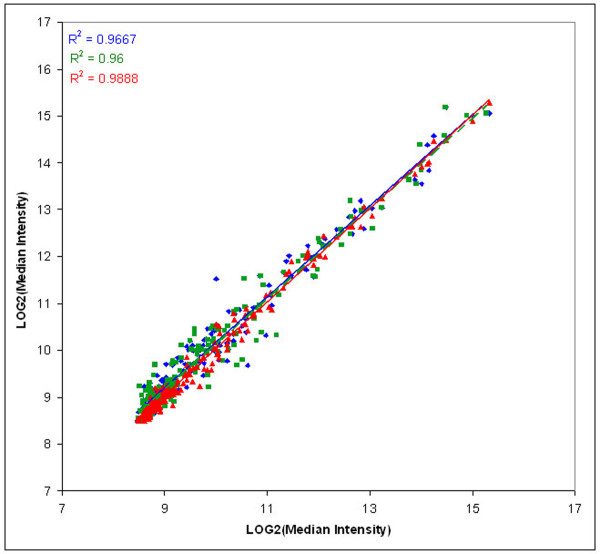
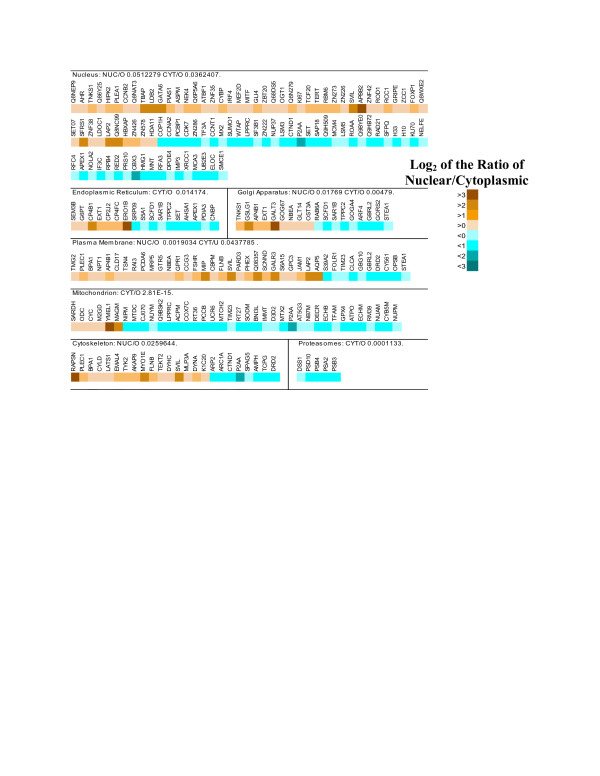
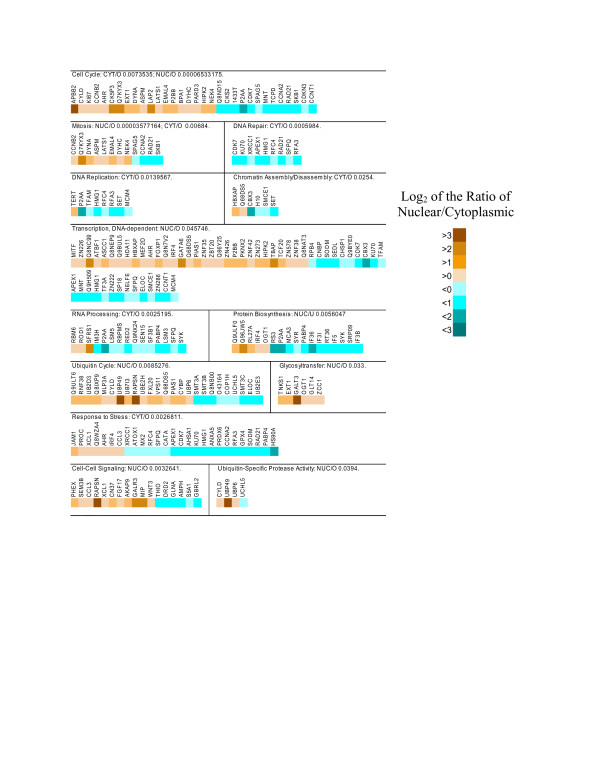
Results: Using microarrays comprising oligonucleotide probes that represent either protein-coding transcripts or microRNAs (miRNA), we have studied global transcript accumulation patterns for the HepG2 (human hepatoma) cell line. Through subdividing the total pool of RNA transcripts into samples from nuclei, the cytoplasm, and whole cells, we determined the degree of correlation of these patterns across these different subcellular locations. The transcript and miRNA abundance patterns for the three RNA fractions were largely similar, but with some exceptions: nuclear RNA samples were enriched with respect to the cytoplasm in transcripts encoding proteins associated with specific nuclear functions, such as the cell cycle, mitosis, and transcription. The cytoplasmic RNA fraction also was enriched, when compared to the nucleus, in transcripts for proteins related to specific nuclear functions, including the cell cycle, DNA replication, and DNA repair. Some transcripts related to the ubiquitin cycle, and transcripts for various membrane proteins were sorted into either the nuclear or cytoplasmic fractions.
Conclusion: Enrichment or compartmentalization of cell cycle and ubiquitin cycle transcripts within the nucleus may be related to the regulation of their expression, by preventing their translation to proteins. In this way, these cellular functions may be tightly controlled by regulating the release of mRNA from the nucleus and thereby the expression of key rate limiting steps in these pathways. Many miRNA precursors were also enriched in the nuclear samples, with significantly fewer being enriched in the cytoplasm. Studies of mRNA localization will help to clarify the roles RNA processing and transport play in the regulation of cellular function.
Figures
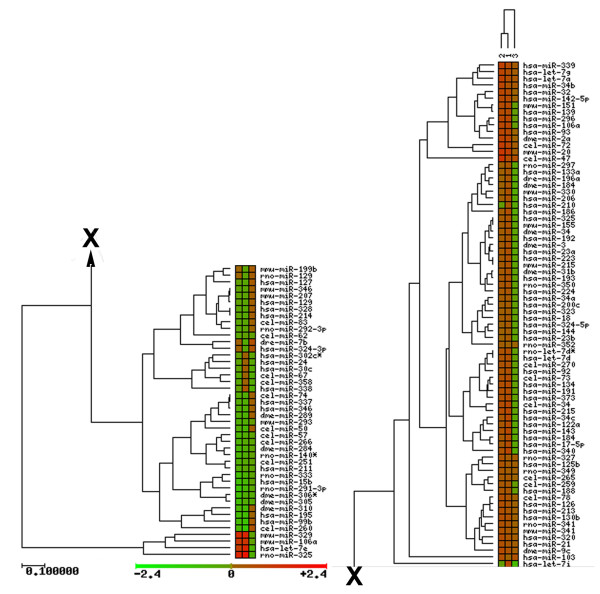
References
-
- Hughes TR, Marton MJ, Jones AR, Roberts CJ, Stoughton R, Armour CD, Bennett HA, Coffey E, Dai H, He YD, Kidd MJ, King AM, Meyer MR, Slade D, Lum PY, Stepaniants SB, Shoemaker DD, Gachotte D, Chakraburtty K, Simon J, Bard M, Friend SH. Functional discovery via a compendium of expression profiles. Cell. 2000;102:109–126. doi: 10.1016/S0092-8674(00)00015-5. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
