

Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia
- PMID: 17898326
- PMCID: PMC4094148
- DOI: 10.1212/01.wnl.0000265397.70057.d8
Cold-induced defects of sodium channel gating in atypical periodic paralysis plus myotonia
Abstract
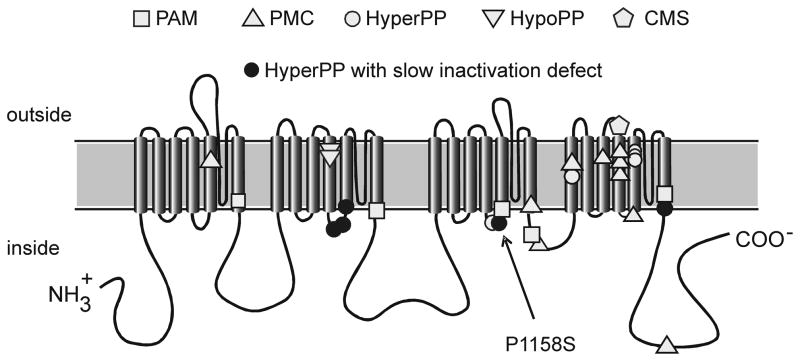
Background: Missense mutations of the skeletal muscle voltage-gated sodium channel (NaV1.4) are an established cause of several clinically distinct forms of periodic paralysis and myotonia. The mechanistic basis for the phenotypic variability of these allelic disorders of muscle excitability remains unknown. An atypical phenotype with cold-induced hypokalemic paralysis and myotonia at warm temperatures was reported to segregate with the P1158S mutation.
Objective: This study extends the functional characterization of the P1158S mutation and tests the specific hypothesis that impairment of Na channel slow inactivation is a common feature of periodic paralysis.
Methods: Mutant NaV1.4 channels (P1158S) were transiently expressed in human embryonic kidney cells and characterized by voltage-clamp studies of Na currents.
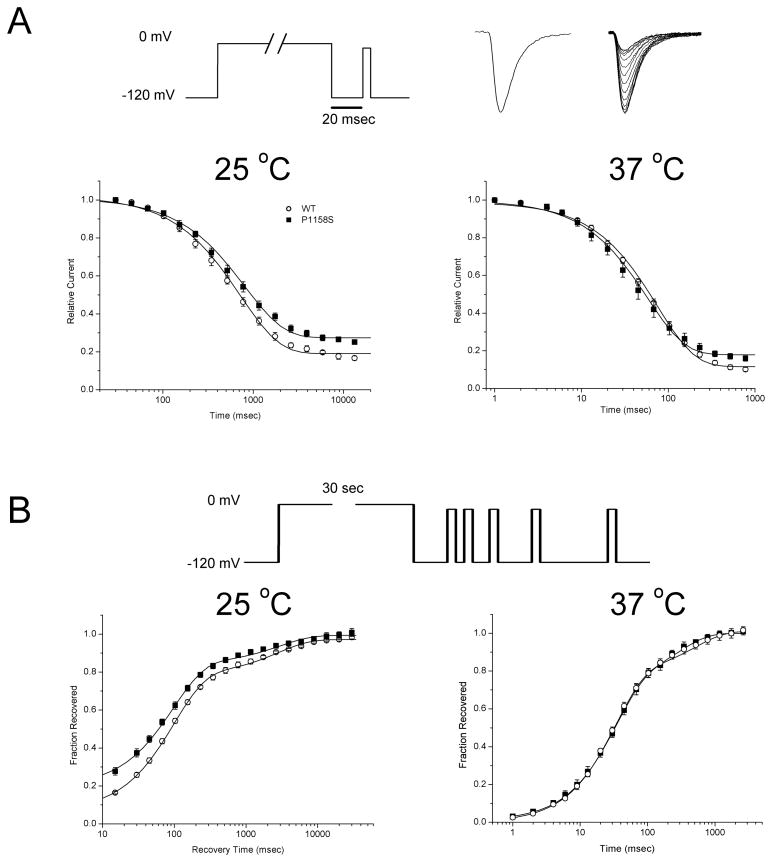
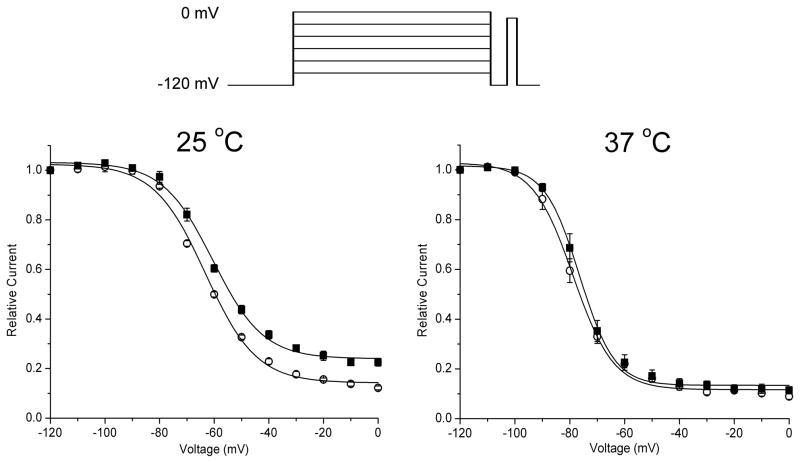
Results: Wild-type and P1158S channels displayed comparable behavior at 37 degrees C, but upon cooling to 25 degrees C, mutant channels activated at more negative potentials and slow inactivation was destabilized.
Conclusions: Consistent with other NaV1.4 mutations associated with a paralytic phenotype, the P1158S mutation disrupts slow inactivation. The unique temperature sensitivity of the channel defect may contribute to the unusual clinical phenotype.
Conflict of interest statement
Disclosure: The authors report no conflict of interest
Figures
Comment in
-
Slow inactivation: slow but not dull.Neurology. 2008 Mar 4;70(10):746-7. doi: 10.1212/01.wnl.0000304253.88642.8d. Neurology. 2008. PMID: 18316687 No abstract available.
References
-
- Cannon SC. Pathomechanisms in channelopathies of skeletal muscle and brain. Annu Rev Neurosci. 2006;29:387–415. - PubMed
-
- Lehmann-Horn F, Jurkat-Rott K. Voltage-gated ion channels and hereditary disease. Physiol Rev. 1999;79:1317–1372. - PubMed
-
- Sugiura Y, Aoki T, Sugiyama Y, Hida C, Ogata M, Yamamoto T. Temperature-sensitive sodium channelopathy with heat-induced myotonia and cold-induced paralysis. Neurology. 2000;54:2179–2181. - PubMed
-
- Gasser T, Dichgans M, Finsterer J, et al. EFNS Task Force on Molecular Diagnosis of Neurologic Disorders: guidelines for the molecular diagnosis of inherited neurologic diseases. Second of two parts. Eur J Neurol. 2001;8:407–424. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases