

Novel and recurrent ALDH3A2 mutations in Italian patients with Sjögren-Larsson syndrome
- PMID: 17902024
- PMCID: PMC3057174
- DOI: 10.1007/s10038-007-0180-z
Novel and recurrent ALDH3A2 mutations in Italian patients with Sjögren-Larsson syndrome
Abstract
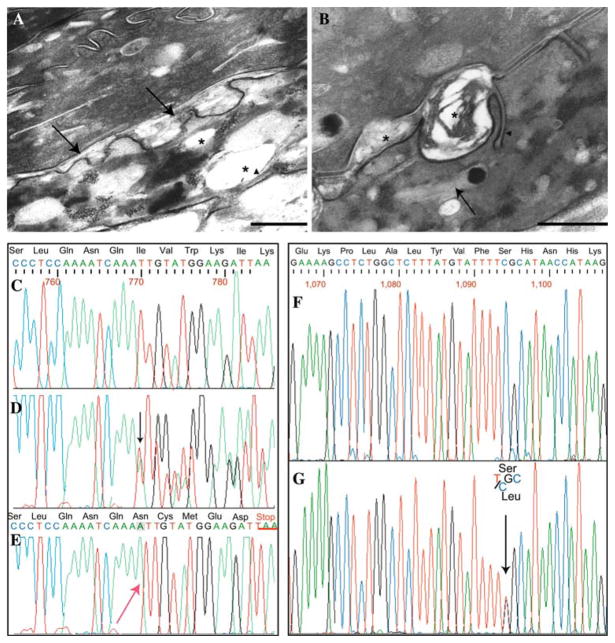
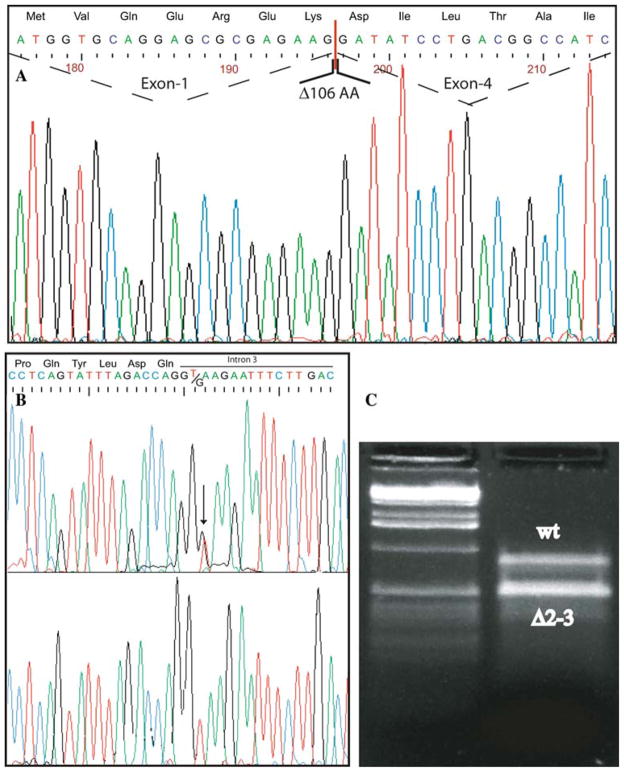
Sjögren-Larsson syndrome (SLS; MIM#270200) is an autosomal recessive neurocutaneous disease caused by mutations in the ALDH3A2 gene for fatty aldehyde dehydrogenase (FALDH), a microsomal enzyme that catalyzes the oxidation of medium- and long- chain aliphatic aldehydes fatty acids. We studied two unrelated Italian SLS patients with ichthyosis, developmental delay, spastic diplegia and brain white matter disease. One patient was homozygous for a novel ALDH3A2 insertion mutation (c.767insA) in exon 5. The other SLS patient was a compound heterozygote for two previously reported mutations: a slice site mutation (c.1094C > T; S365L) in exon 7. Analysis of fibroblast RNA by RT-PCR indicated that the spice-site mutation caused skipping of exons 2 and 3. The c.1094C > T mutation, previously associated with two ALDH3A2 haplotypes, was found on a third distinct haplotype in our patient, which indicates that arose independently in this kindred. These results add to understanding of the genetic basis of SLS and will be useful for DNA diagnosis of this disease.
Figures
References
-
- Bernardini ML, Cangiotti AM, Zamponi N, Porfiri L, Cinti S, Offidani A. Diagnosing Sjogren–Larsson syndrome in a 7-year-old Moroccan boy. J Cutan Pathol. 2007;34:270–275. - PubMed
-
- Chang C, Yoshida A. Human fatty aldehyde dehydrogenase gene (ALDH10): organization and tissue-dependent expression. Genomics. 1997;40:80–85. - PubMed
-
- De Laurenzi V, Rogers GR, Hamrock DJ, Marekov LN, Steinert PM, Compton JG, Markova N, Rizzo WB. Sjogren–Larsson syndrome is caused by mutations in the fatty aldehyde dehydrogenase gene. Nat Genet. 1996;12:52–57. - PubMed
-
- Hempel J, Kuo I, Perozich J, Wang BC, Lindahl R, Nicholas H. Aldehyde dehydrogenase. Maintaining critical active site geometry at motif 8 in the class 3 enzyme. Eur J Biochem. 2001a;268:722–726. - PubMed
-
- Hempel J, Lindahl R, Perozich J, Wang B, Kuo I, Nicholas H. Beyond the catalytic core of ALDH: a web of important residues begins to emerge. Chem Biol Interact. 2001b;130–132:39–46. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources