

Finding regulatory elements and regulatory motifs: a general probabilistic framework
- PMID: 17903285
- PMCID: PMC1995539
- DOI: 10.1186/1471-2105-8-S6-S4
Finding regulatory elements and regulatory motifs: a general probabilistic framework
Abstract
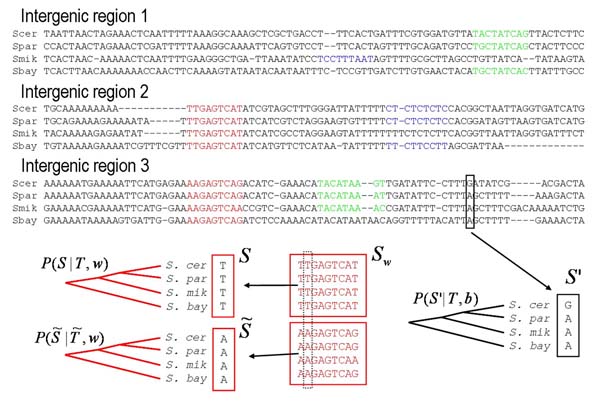
Over the last two decades a large number of algorithms has been developed for regulatory motif finding. Here we show how many of these algorithms, especially those that model binding specificities of regulatory factors with position specific weight matrices (WMs), naturally arise within a general Bayesian probabilistic framework. We discuss how WMs are constructed from sets of regulatory sites, how sites for a given WM can be discovered by scanning of large sequences, how to cluster WMs, and more generally how to cluster large sets of sites from different WMs into clusters. We discuss how 'regulatory modules', clusters of sites for subsets of WMs, can be found in large intergenic sequences, and we discuss different methods for ab initio motif finding, including expectation maximization (EM) algorithms, and motif sampling algorithms. Finally, we extensively discuss how module finding methods and ab initio motif finding methods can be extended to take phylogenetic relations between the input sequences into account, i.e. we show how motif finding and phylogenetic footprinting can be integrated in a rigorous probabilistic framework. The article is intended for readers with a solid background in applied mathematics, and preferably with some knowledge of general Bayesian probabilistic methods. The main purpose of the article is to elucidate that all these methods are not a disconnected set of individual algorithmic recipes, but that they are just different facets of a single integrated probabilistic theory.
Figures








References
-
- Roulet E, Busso S, Camargo AA, Simpson AJ, Mermod N, Bucher P. High-throughput SELEX-SAGE method for quantitative modeling of transcription-factor binding sites. Nat Biotechnol. 2002;20:831–835. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
