

Toward high-resolution prediction and design of transmembrane helical protein structures
- PMID: 17905872
- PMCID: PMC2000396
- DOI: 10.1073/pnas.0702515104
Toward high-resolution prediction and design of transmembrane helical protein structures
Erratum in
- Proc Natl Acad Sci U S A. 2007 Dec 18;104(51):20635
Abstract
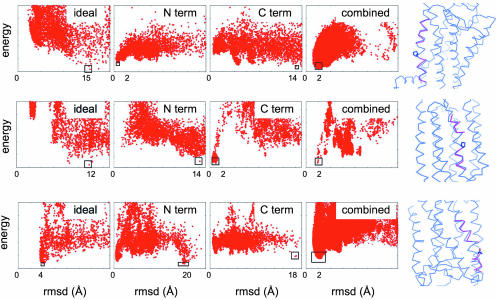
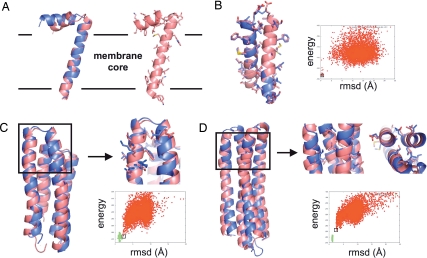
The prediction and design at the atomic level of membrane protein structures and interactions is a critical but unsolved challenge. To address this problem, we have developed an all-atom physical model that describes intraprotein and protein-solvent interactions in the membrane environment. We evaluated the ability of the model to recapitulate the energetics and structural specificities of polytopic membrane proteins by using a battery of in silico prediction and design tests. First, in side-chain packing and design tests, the model successfully predicts the side-chain conformations at 73% of nonexposed positions and the native amino acid identities at 34% of positions in naturally occurring membrane proteins. Second, the model predicts significant energy gaps between native and nonnative structures of transmembrane helical interfaces and polytopic membrane proteins. Third, distortions in transmembrane helices are successfully recapitulated in docking experiments by using fragments of ideal helices judiciously defined around helical kinks. Finally, de novo structure prediction reaches near-atomic accuracy (<2.5 A) for several small membrane protein domains (<150 residues). The success of the model highlights the critical role of van der Waals and hydrogen-bonding interactions in the stability and structural specificity of membrane protein structures and sets the stage for the high-resolution prediction and design of complex membrane protein architectures.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
