

Members of a large retroposon family are determinants of post-transcriptional gene expression in Leishmania
- PMID: 17907803
- PMCID: PMC2323293
- DOI: 10.1371/journal.ppat.0030136
Members of a large retroposon family are determinants of post-transcriptional gene expression in Leishmania
Abstract
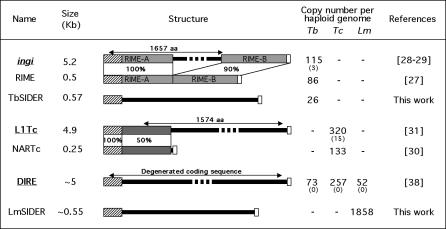
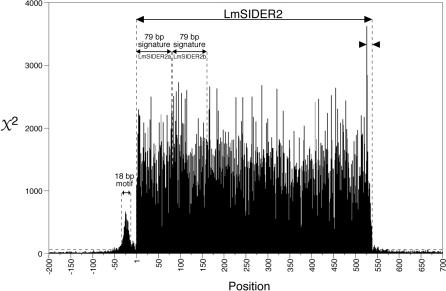
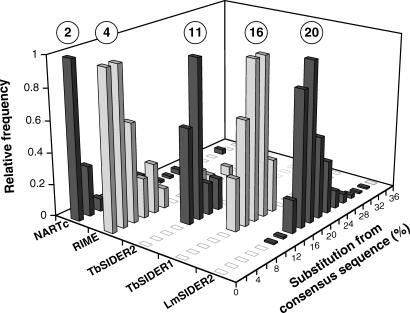
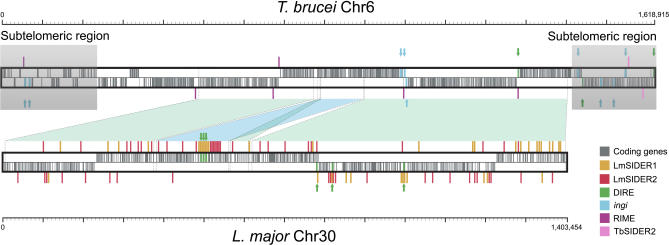
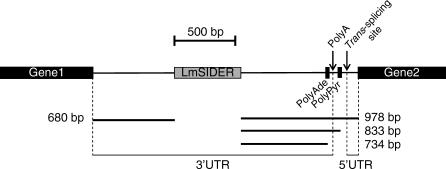
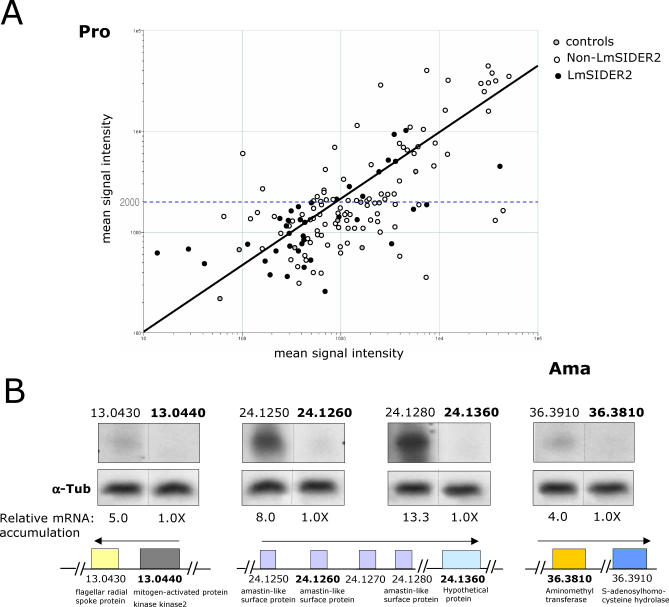
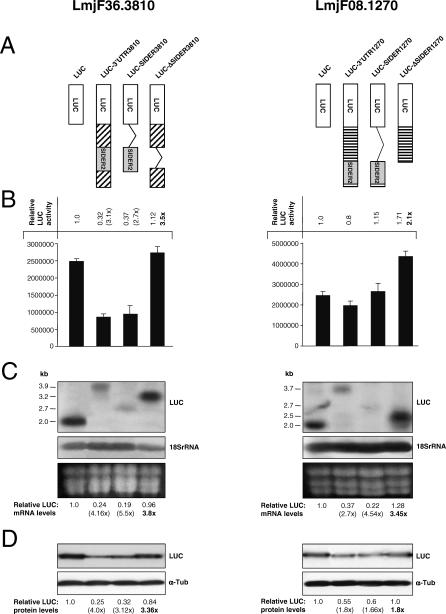
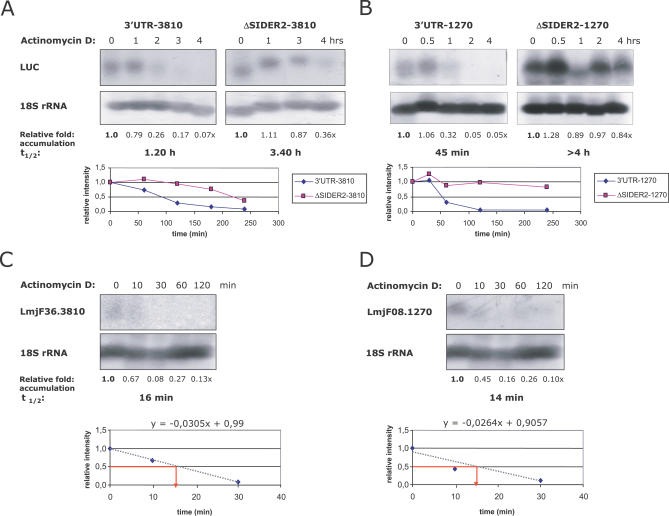
Trypanosomatids are unicellular protists that include the human pathogens Leishmania spp. (leishmaniasis), Trypanosoma brucei (sleeping sickness), and Trypanosoma cruzi (Chagas disease). Analysis of their recently completed genomes confirmed the presence of non-long-terminal repeat retrotransposons, also called retroposons. Using the 79-bp signature sequence common to all trypanosomatid retroposons as bait, we identified in the Leishmania major genome two new large families of small elements--LmSIDER1 (785 copies) and LmSIDER2 (1,073 copies)--that fulfill all the characteristics of extinct trypanosomatid retroposons. LmSIDERs are approximately 70 times more abundant in L. major compared to T. brucei and are found almost exclusively within the 3'-untranslated regions (3'UTRs) of L. major mRNAs. We provide experimental evidence that LmSIDER2 act as mRNA instability elements and that LmSIDER2-containing mRNAs are generally expressed at lower levels compared to the non-LmSIDER2 mRNAs. The considerable expansion of LmSIDERs within 3'UTRs in an organism lacking transcriptional control and their role in regulating mRNA stability indicate that Leishmania have probably recycled these short retroposons to globally modulate the expression of a number of genes. To our knowledge, this is the first example in eukaryotes of the domestication and expansion of a family of mobile elements that have evolved to fulfill a critical cellular function.
Conflict of interest statement
Figures

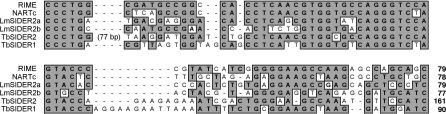

References
-
- Haag J, O'Huigin C, Overath P. The molecular phylogeny of trypanosomes: Evidence for an early divergence of the Salivaria. Mol Biochem Parasitol. 1998;91:37–49. - PubMed
-
- Berriman M, Ghedin E, Hertz-Fowler C, Blandin G, Renauld H, et al. The genome of the African trypanosome Trypanosoma brucei. Science. 2005;309:416–422. - PubMed
-
- El-Sayed NM, Myler PJ, Bartholomeu DC, Nilsson D, Aggarwal G, et al. The genome sequence of Trypanosoma cruzi, etiologic agent of Chagas disease. Science. 2005;309:409–415. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
