

SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing
- PMID: 17908823
- PMCID: PMC2045152
- DOI: 10.1101/gr.6435207
SHARCGS, a fast and highly accurate short-read assembly algorithm for de novo genomic sequencing
Abstract
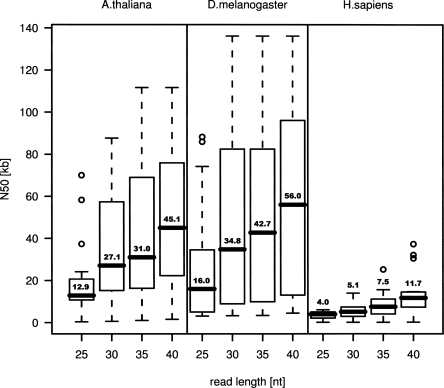
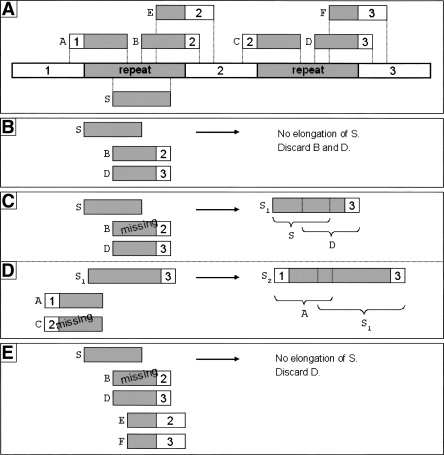
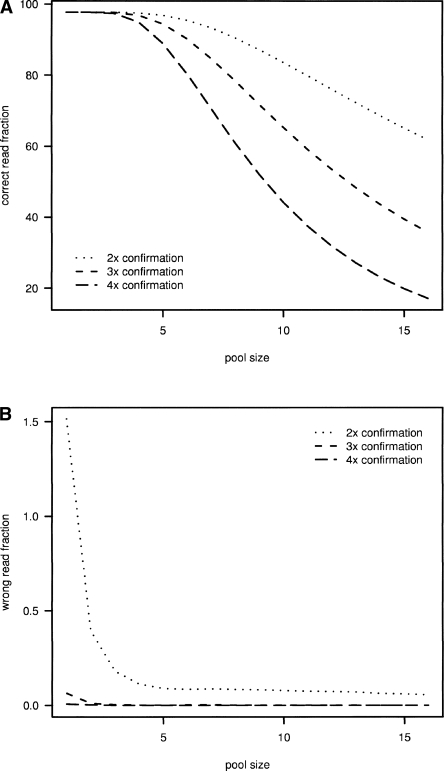
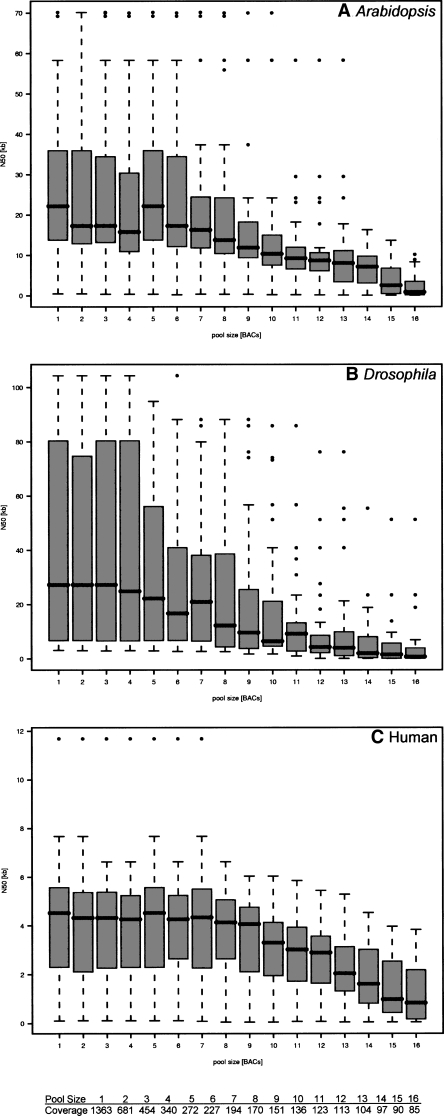
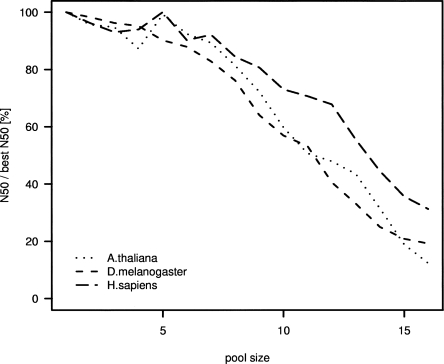
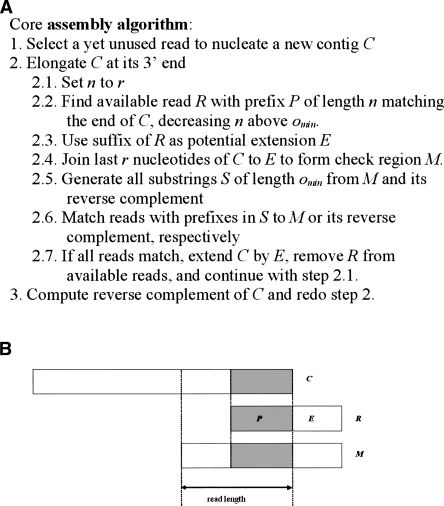
The latest revolution in the DNA sequencing field has been brought about by the development of automated sequencers that are capable of generating giga base pair data sets quickly and at low cost. Applications of such technologies seem to be limited to resequencing and transcript discovery, due to the shortness of the generated reads. In order to extend the fields of application to de novo sequencing, we developed the SHARCGS algorithm to assemble short-read (25-40-mer) data with high accuracy and speed. The efficiency of SHARCGS was tested on BAC inserts from three eukaryotic species, on two yeast chromosomes, and on two bacterial genomes (Haemophilus influenzae, Escherichia coli). We show that 30-mer-based BAC assemblies have N50 sizes >20 kbp for Drosophila and Arabidopsis and >4 kbp for human in simulations taking missing reads and wrong base calls into account. We assembled 949,974 contigs with length >50 bp, and only one single contig could not be aligned error-free against the reference sequences. We generated 36-mer reads for the genome of Helicobacter acinonychis on the Illumina 1G sequencing instrument and assembled 937 contigs covering 98% of the genome with an N50 size of 3.7 kbp. With the exception of five contigs that differ in 1-4 positions relative to the reference sequence, all contigs matched the genome error-free. Thus, SHARCGS is a suitable tool for fully exploiting novel sequencing technologies by assembling sequence contigs de novo with high confidence and by outperforming existing assembly algorithms in terms of speed and accuracy.
Figures
References
-
- Adams M.D., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Celniker S.E., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Holt R.A., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Evans C.A., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Gocayne J.D., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Amanatides P.G., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Scherer S.E., Li P.W., Hoskins R.A., Galle R.F., Li P.W., Hoskins R.A., Galle R.F., Hoskins R.A., Galle R.F., Galle R.F. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. - PubMed
-
- Aparicio S., Chapman J., Stupka E., Putnam N., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Chapman J., Stupka E., Putnam N., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Stupka E., Putnam N., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Putnam N., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Chia J.M., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Dehal P., Christoffels A., Rash S., Hoon S., Smit A., Christoffels A., Rash S., Hoon S., Smit A., Rash S., Hoon S., Smit A., Hoon S., Smit A., Smit A., et al. Whole-genome shotgun assembly and analysis of the genome of Fugu rubripes. Science. 2002;297:1301–1310. - PubMed
-
- Batzoglou S., Jaffe D.B., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Jaffe D.B., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Stanley K., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Butler J., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Gnerre S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Mauceli E., Berger B., Mesirov J.P., Lander E.S., Berger B., Mesirov J.P., Lander E.S., Mesirov J.P., Lander E.S., Lander E.S. ARACHNE: A whole-genome shotgun assembler. Genome Res. 2002;12:177–189. doi: 10.1101/gr.208902. - DOI - PMC - PubMed
-
- Bentley D.R. Whole-genome re-sequencing. Curr. Opin. Genet. Dev. 2006;16:545–552. - PubMed
-
- Choi S.D., Creelman R., Mullet J., Wing R.A., Creelman R., Mullet J., Wing R.A., Mullet J., Wing R.A., Wing R.A. Construction and characterization of a bacterial artificial chromosome library from Arabidopsis thaliana. Weeds World. 1995;2:17–20.
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous