

A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease
- PMID: 17908927
- PMCID: PMC1993871
- DOI: 10.1101/gad.1553207
A role for AGL ubiquitination in the glycogen storage disorders of Lafora and Cori's disease
Abstract
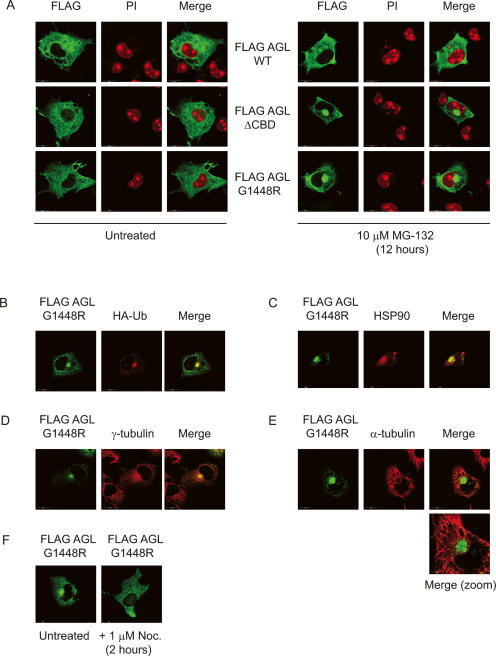
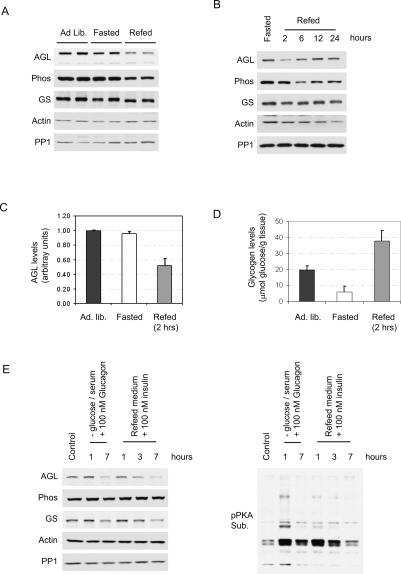
Cori's disease is a glycogen storage disorder characterized by a deficiency in the glycogen debranching enzyme, amylo-1,6-glucosidase,4-alpha-glucanotransferase (AGL). Here, we demonstrate that the G1448R genetic variant of AGL is unable to bind to glycogen and displays decreased stability that is rescued by proteasomal inhibition. AGL G1448R is more highly ubiquitinated than its wild-type counterpart and forms aggresomes upon proteasome impairment. Furthermore, the E3 ubiquitin ligase Malin interacts with and promotes the ubiquitination of AGL. Malin is known to be mutated in Lafora disease, an autosomal recessive disorder clinically characterized by the accumulation of polyglucosan bodies resembling poorly branched glycogen. Transfection studies in HepG2 cells demonstrate that AGL is cytoplasmic whereas Malin is predominately nuclear. However, after depletion of glycogen stores for 4 h, approximately 90% of transfected cells exhibit partial nuclear staining for AGL. Furthermore, stimulation of cells with agents that elevate cAMP increases Malin levels and Malin/AGL complex formation. Refeeding mice for 2 h after an overnight fast causes a reduction in hepatic AGL levels by 48%. Taken together, these results indicate that binding to glycogen crucially regulates the stability of AGL and, further, that its ubiquitination may play an important role in the pathophysiology of both Lafora and Cori's disease.
Figures





Similar articles
-
Laforin-malin complex degrades polyglucosan bodies in concert with glycogen debranching enzyme and brain isoform glycogen phosphorylase.Mol Neurobiol. 2014 Apr;49(2):645-57. doi: 10.1007/s12035-013-8546-z. Epub 2013 Sep 26. Mol Neurobiol. 2014. PMID: 24068615 Free PMC article.
-
Distinct mutations in the glycogen debranching enzyme found in glycogen storage disease type III lead to impairment in diverse cellular functions.Hum Mol Genet. 2009 Jun 1;18(11):2045-52. doi: 10.1093/hmg/ddp128. Epub 2009 Mar 19. Hum Mol Genet. 2009. PMID: 19299494 Free PMC article.
-
Co-chaperone CHIP stabilizes aggregate-prone malin, a ubiquitin ligase mutated in Lafora disease.J Biol Chem. 2010 Jan 8;285(2):1404-13. doi: 10.1074/jbc.M109.006312. Epub 2009 Nov 5. J Biol Chem. 2010. PMID: 19892702 Free PMC article.
-
Advances in lafora progressive myoclonus epilepsy.Curr Neurol Neurosci Rep. 2007 Sep;7(5):428-33. doi: 10.1007/s11910-007-0066-7. Curr Neurol Neurosci Rep. 2007. PMID: 17764634 Review.
-
Deciphering the role of malin in the lafora progressive myoclonus epilepsy.IUBMB Life. 2012 Oct;64(10):801-8. doi: 10.1002/iub.1072. Epub 2012 Jul 20. IUBMB Life. 2012. PMID: 22815132 Free PMC article. Review.
Cited by
-
Lafora disease offers a unique window into neuronal glycogen metabolism.J Biol Chem. 2018 May 11;293(19):7117-7125. doi: 10.1074/jbc.R117.803064. Epub 2018 Feb 26. J Biol Chem. 2018. PMID: 29483193 Free PMC article. Review.
-
The dynamic life of the glycogen granule.J Biol Chem. 2018 May 11;293(19):7089-7098. doi: 10.1074/jbc.R117.802843. Epub 2018 Feb 26. J Biol Chem. 2018. PMID: 29483195 Free PMC article. Review.
-
AMP-activated protein kinase phosphorylates R5/PTG, the glycogen targeting subunit of the R5/PTG-protein phosphatase 1 holoenzyme, and accelerates its down-regulation by the laforin-malin complex.J Biol Chem. 2009 Mar 27;284(13):8247-55. doi: 10.1074/jbc.M808492200. Epub 2009 Jan 26. J Biol Chem. 2009. PMID: 19171932 Free PMC article.
-
Laforin, a dual specificity phosphatase involved in Lafora disease, is present mainly as monomeric form with full phosphatase activity.PLoS One. 2011;6(8):e24040. doi: 10.1371/journal.pone.0024040. Epub 2011 Aug 26. PLoS One. 2011. PMID: 21887368 Free PMC article.
-
The role of ubiquitination and deubiquitination in cancer metabolism.Mol Cancer. 2020 Oct 1;19(1):146. doi: 10.1186/s12943-020-01262-x. Mol Cancer. 2020. PMID: 33004065 Free PMC article. Review.
References
-
- Brady M.J., Saltiel A.R., Saltiel A.R. The role of protein phosphatase-1 in insulin action. Recent Prog. Horm. Res. 2001;56:157–173. - PubMed
-
- Chan E.M., Young E.J., Ianzano L., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Young E.J., Ianzano L., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Ianzano L., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Munteanu I., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Zhao X., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Christopoulos C.C., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Avanzini G., Elia M., Ackerley C.A., Jovic N.J., Elia M., Ackerley C.A., Jovic N.J., Ackerley C.A., Jovic N.J., Jovic N.J., et al. Mutations in NHLRC1 cause progressive myoclonus epilepsy. Nat. Genet. 2003;35:125–127. - PubMed
-
- Chan E.M., Ackerley C.A., Lohi H., Ianzano L., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A., Ackerley C.A., Lohi H., Ianzano L., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A., Lohi H., Ianzano L., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A., Ianzano L., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A., Cortez M.A., Shannon P., Scherer S.W., Minassian B.A., Shannon P., Scherer S.W., Minassian B.A., Scherer S.W., Minassian B.A., Minassian B.A. Laforin preferentially binds the neurotoxic starch-like polyglucosans, which form in its absence in progressive myoclonus epilepsy. Hum. Mol. Genet. 2004;13:1117–1129. - PubMed
-
- Chan E.M., Andrade D.M., Franceschetti S., Minassian B., Andrade D.M., Franceschetti S., Minassian B., Franceschetti S., Minassian B., Minassian B. Progressive myoclonus epilepsies: EPM1, EPM2A, EPM2B. Adv. Neurol. 2005;95:47–57. - PubMed
-
- Crosson S.M., Khan A., Printen J., Pessin J.E., Saltiel A.R., Khan A., Printen J., Pessin J.E., Saltiel A.R., Printen J., Pessin J.E., Saltiel A.R., Pessin J.E., Saltiel A.R., Saltiel A.R. PTG gene deletion causes impaired glycogen synthesis and developmental insulin resistance. J. Clin. Invest. 2003;111:1423–1432. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases