

Antibody-protein interactions: benchmark datasets and prediction tools evaluation
- PMID: 17910770
- PMCID: PMC2174481
- DOI: 10.1186/1472-6807-7-64
Antibody-protein interactions: benchmark datasets and prediction tools evaluation
Abstract
Background: The ability to predict antibody binding sites (aka antigenic determinants or B-cell epitopes) for a given protein is a precursor to new vaccine design and diagnostics. Among the various methods of B-cell epitope identification X-ray crystallography is one of the most reliable methods. Using these experimental data computational methods exist for B-cell epitope prediction. As the number of structures of antibody-protein complexes grows, further interest in prediction methods using 3D structure is anticipated. This work aims to establish a benchmark for 3D structure-based epitope prediction methods.
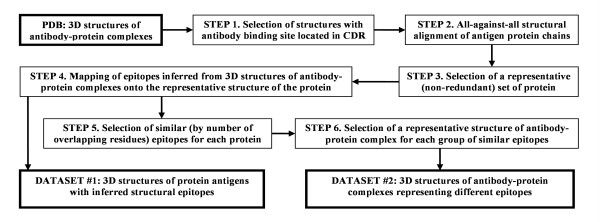
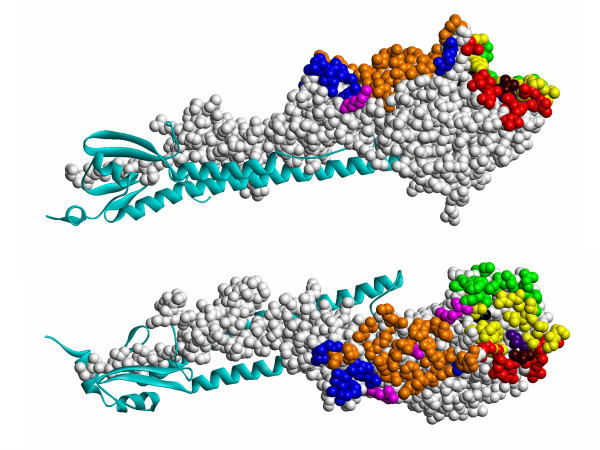
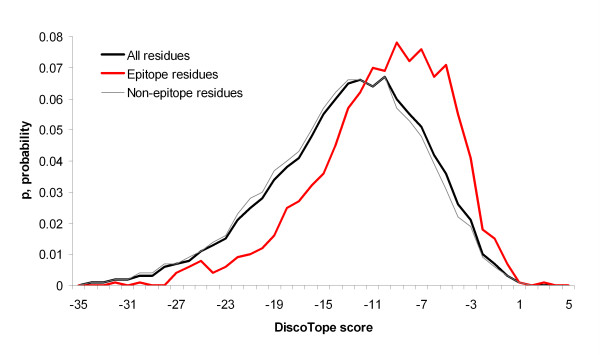
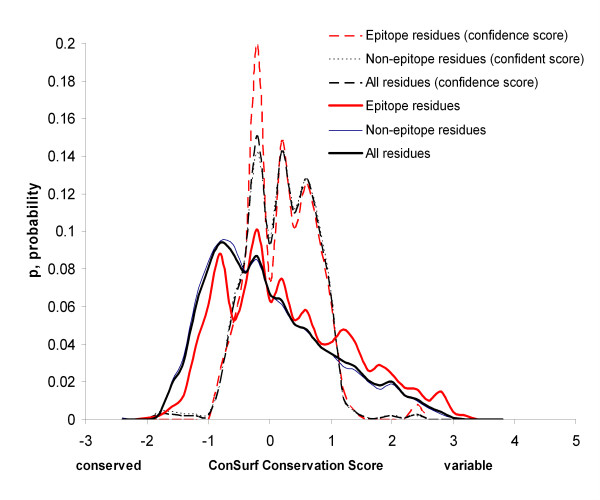
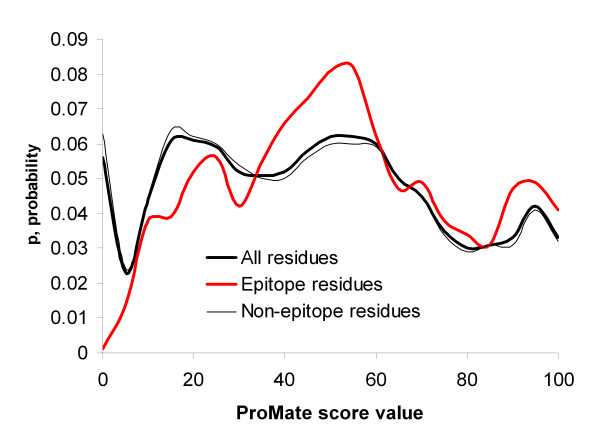
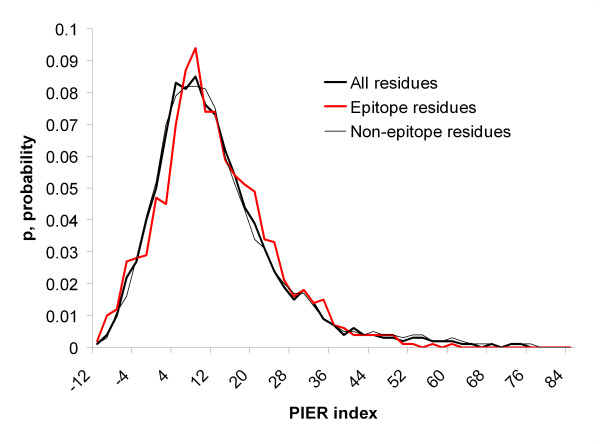
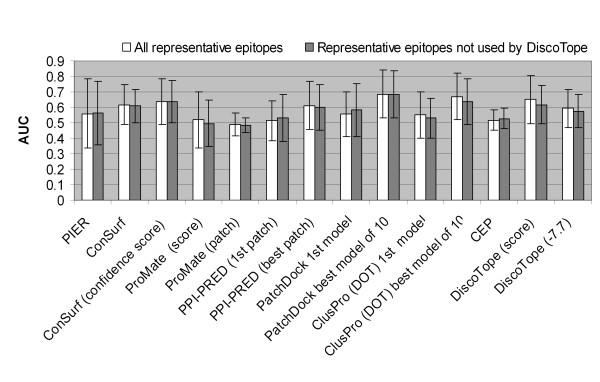
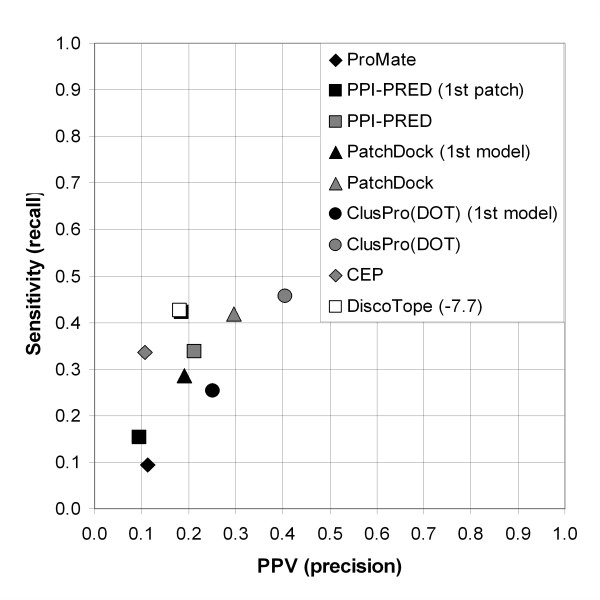
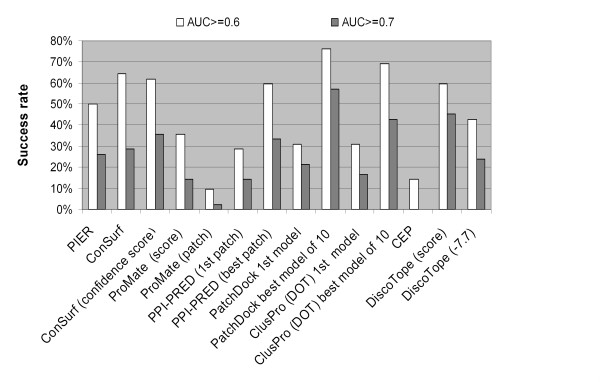
Results: Two B-cell epitope benchmark datasets inferred from the 3D structures of antibody-protein complexes were defined. The first is a dataset of 62 representative 3D structures of protein antigens with inferred structural epitopes. The second is a dataset of 82 structures of antibody-protein complexes containing different structural epitopes. Using these datasets, eight web-servers developed for antibody and protein binding sites prediction have been evaluated. In no method did performance exceed a 40% precision and 46% recall. The values of the area under the receiver operating characteristic curve for the evaluated methods were about 0.6 for ConSurf, DiscoTope, and PPI-PRED methods and above 0.65 but not exceeding 0.70 for protein-protein docking methods when the best of the top ten models for the bound docking were considered; the remaining methods performed close to random. The benchmark datasets are included as a supplement to this paper.
Conclusion: It may be possible to improve epitope prediction methods through training on datasets which include only immune epitopes and through utilizing more features characterizing epitopes, for example, the evolutionary conservation score. Notwithstanding, overall poor performance may reflect the generality of antigenicity and hence the inability to decipher B-cell epitopes as an intrinsic feature of the protein. It is an open question as to whether ultimately discriminatory features can be found.
Figures

Similar articles
-
EPSVR and EPMeta: prediction of antigenic epitopes using support vector regression and multiple server results.BMC Bioinformatics. 2010 Jul 16;11:381. doi: 10.1186/1471-2105-11-381. BMC Bioinformatics. 2010. PMID: 20637083 Free PMC article.
-
ElliPro: a new structure-based tool for the prediction of antibody epitopes.BMC Bioinformatics. 2008 Dec 2;9:514. doi: 10.1186/1471-2105-9-514. BMC Bioinformatics. 2008. PMID: 19055730 Free PMC article.
-
Prediction of conformational B-cell epitopes from 3D structures by random forests with a distance-based feature.BMC Bioinformatics. 2011 Aug 17;12:341. doi: 10.1186/1471-2105-12-341. BMC Bioinformatics. 2011. PMID: 21846404 Free PMC article.
-
Bioinformatics resources and tools for conformational B-cell epitope prediction.Comput Math Methods Med. 2013;2013:943636. doi: 10.1155/2013/943636. Epub 2013 Jul 21. Comput Math Methods Med. 2013. PMID: 23970944 Free PMC article. Review.
-
An Introduction to B-Cell Epitope Mapping and In Silico Epitope Prediction.J Immunol Res. 2016;2016:6760830. doi: 10.1155/2016/6760830. Epub 2016 Dec 29. J Immunol Res. 2016. PMID: 28127568 Free PMC article. Review.
Cited by
-
B-Pred, a structure based B-cell epitopes prediction server.Adv Appl Bioinform Chem. 2012;5:11-21. doi: 10.2147/AABC.S30620. Epub 2012 Jul 25. Adv Appl Bioinform Chem. 2012. PMID: 22888263 Free PMC article.
-
An analysis of B-cell epitope discontinuity.Mol Immunol. 2012 Jul;51(3-4):304-9. doi: 10.1016/j.molimm.2012.03.030. Epub 2012 Apr 20. Mol Immunol. 2012. PMID: 22520973 Free PMC article.
-
Validation of glypican-3-specific scFv isolated from paired display/secretory yeast display library.BMC Biotechnol. 2012 May 7;12:23. doi: 10.1186/1472-6750-12-23. BMC Biotechnol. 2012. PMID: 22564378 Free PMC article.
-
Epitope prediction based on random peptide library screening: benchmark dataset and prediction tools evaluation.Molecules. 2011 Jun 16;16(6):4971-93. doi: 10.3390/molecules16064971. Molecules. 2011. PMID: 21681149 Free PMC article.
-
A review of quantitative modeling of B cell responses to antigenic challenge.J Pharmacokinet Pharmacodyn. 2014 Oct;41(5):445-59. doi: 10.1007/s10928-014-9388-7. Epub 2014 Oct 19. J Pharmacokinet Pharmacodyn. 2014. PMID: 25326873 Review.
References
-
- Peters B, Sidney J, Bourne P, Bui HH, Buus S, Doh G, Fleri W, Kronenberg M, Kubo R, Lund O, Nemazee D, Ponomarenko JV, Sathiamurthy M, Schoenberger SP, Stewart S, Surko P, Way S, Wilson S, Sette A. The design and implementation of the immune epitope database and analysis resource. Immunogenetics. 2005;57:326–336. - PMC - PubMed
-
- Van Regenmortel MH. Immunoinformatics may lead to a reappraisal of the nature of B cell epitopes and of the feasibility of synthetic peptide vaccines. J Mol Recognit. 2006;19:183–187. - PubMed
-
- Gomara MJ, Haro I. Synthetic peptides for the immunodiagnosis of human diseases. Curr Med Chem. 2007;14:531–546. - PubMed
-
- Meloen RH, Puijk WC, Langeveld JP, Langedijk JP, Timmerman P. Design of synthetic peptides for diagnostics. Curr Protein Pept Sci. 2003;4:253–260. - PubMed
-
- Leinikki P, Lehtinen M, Hyoty H, Parkkonen P, Kantanen ML, Hakulinen J. Synthetic peptides as diagnostic tools in virology. Adv Virus Res. 1993;42:149–186. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
