

Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death
- PMID: 17911251
- PMCID: PMC2042206
- DOI: 10.1073/pnas.0706662104
Gambogic amide, a selective agonist for TrkA receptor that possesses robust neurotrophic activity, prevents neuronal cell death
Abstract
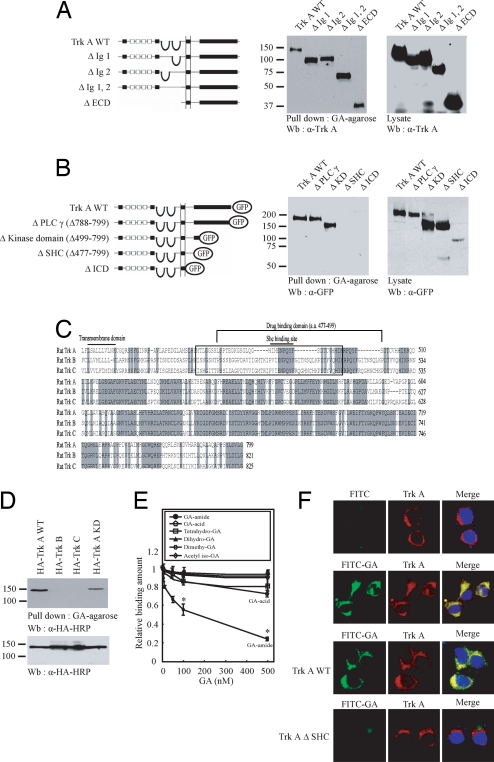
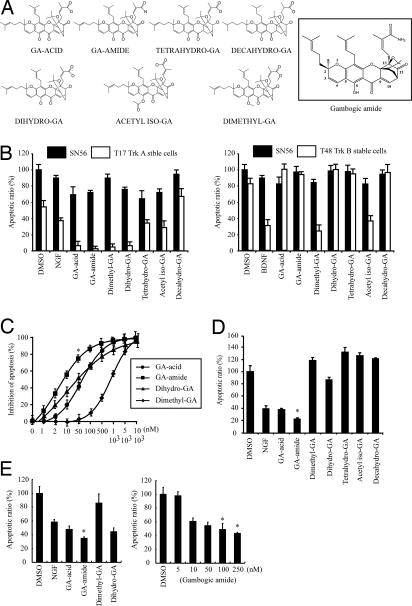
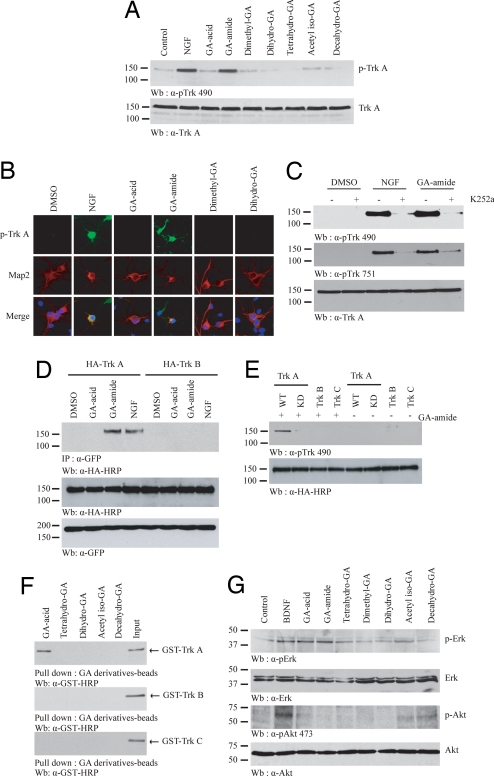
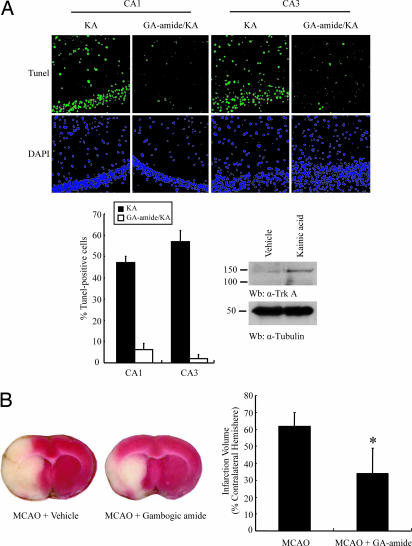
Nerve growth factor (NGF) binds to TrkA receptor and triggers activation of numerous signaling cascades, which play critical roles in neuronal plasticity, survival, and neurite outgrowth. To mimic NGF functions pharmacologically, we developed a high-throughput screening assay to identify small-molecule agonists for TrkA receptor. The most potent compound, gambogic amide, selectively binds to TrkA, but not TrkB or TrkC, and robustly induces its tyrosine phosphorylation and downstream signaling activation, including Akt and MAPKs. Further, it strongly prevents glutamate-induced neuronal cell death and provokes prominent neurite outgrowth in PC12 cells. Gambogic amide specifically interacts with the cytoplasmic juxtamembrane domain of TrkA receptor and triggers its dimerization. Administration of this molecule in mice substantially diminishes kainic acid-triggered neuronal cell death and decreases infarct volume in the transient middle cerebral artery occlusion model of stroke. Thus, gambogic amide might not only establish a powerful platform for dissection of the physiological roles of NGF and TrkA receptor but also provide effective treatments for neurodegenerative diseases and stroke.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Gambogic amide selectively upregulates TrkA expression and triggers its activation.Pharmacol Rep. 2015 Apr;67(2):217-23. doi: 10.1016/j.pharep.2014.09.002. Epub 2014 Sep 22. Pharmacol Rep. 2015. PMID: 25712642
-
Amitriptyline is a TrkA and TrkB receptor agonist that promotes TrkA/TrkB heterodimerization and has potent neurotrophic activity.Chem Biol. 2009 Jun 26;16(6):644-56. doi: 10.1016/j.chembiol.2009.05.010. Chem Biol. 2009. PMID: 19549602 Free PMC article.
-
Functional expression of TrkA receptors in hippocampal neurons.J Neurosci Res. 1998 Nov 1;54(3):424-31. doi: 10.1002/(SICI)1097-4547(19981101)54:3<424::AID-JNR13>3.0.CO;2-6. J Neurosci Res. 1998. PMID: 9819147
-
NGF and ProNGF: Regulation of neuronal and neoplastic responses through receptor signaling.Adv Biol Regul. 2015 May;58:16-27. doi: 10.1016/j.jbior.2014.11.003. Epub 2014 Nov 20. Adv Biol Regul. 2015. PMID: 25491371 Free PMC article. Review.
-
Nerve growth factor-independent neuronal survival: a role for NO donors.Mol Pharmacol. 2005 Oct;68(4):952-5. doi: 10.1124/mol.105.017277. Epub 2005 Jul 26. Mol Pharmacol. 2005. PMID: 16046659 Review.
Cited by
-
TrkA+ Neurons Induce Pathologic Regeneration After Soft Tissue Trauma.Stem Cells Transl Med. 2022 Nov 18;11(11):1165-1176. doi: 10.1093/stcltm/szac073. Stem Cells Transl Med. 2022. PMID: 36222619 Free PMC article.
-
A selective TrkB agonist with potent neurotrophic activities by 7,8-dihydroxyflavone.Proc Natl Acad Sci U S A. 2010 Feb 9;107(6):2687-92. doi: 10.1073/pnas.0913572107. Epub 2010 Jan 25. Proc Natl Acad Sci U S A. 2010. PMID: 20133810 Free PMC article.
-
Transcriptional dysregulation of TrkA associates with neurodegeneration in spinocerebellar ataxia type 17.Hum Mol Genet. 2009 Nov 1;18(21):4141-52. doi: 10.1093/hmg/ddp363. Epub 2009 Jul 30. Hum Mol Genet. 2009. PMID: 19643914 Free PMC article.
-
Natural Products for Neurodegeneration: Regulating Neurotrophic Signals.Oxid Med Cell Longev. 2021 Jun 21;2021:8820406. doi: 10.1155/2021/8820406. eCollection 2021. Oxid Med Cell Longev. 2021. Retraction in: Oxid Med Cell Longev. 2024 Jan 9;2024:9854929. doi: 10.1155/2024/9854929. PMID: 34239696 Free PMC article. Retracted. Review.
-
Recent research on bioactive xanthones from natural medicine: Garcinia hanburyi.AAPS PharmSciTech. 2015 Aug;16(4):742-58. doi: 10.1208/s12249-015-0339-4. Epub 2015 Jul 8. AAPS PharmSciTech. 2015. PMID: 26152816 Free PMC article. Review.
References
-
- Kaplan DR, Stephens RM. J Neurobiol. 1994;25:1404–1417. - PubMed
-
- Mufson EJ, Lavine N, Jaffar S, Kordower JH, Quirion R, Saragovi HU. Exp Neurol. 1997;146:91–103. - PubMed
-
- Counts SE, Nadeem M, Wuu J, Ginsberg SD, Saragovi HU, Mufson EJ. Ann Neurol. 2004;56:520–531. - PubMed
-
- Backman C, Rose GM, Bartus RT, Hoffer BJ, Mufson EJ, Granholm AC. J Comp Neurol. 1997;387:1–11. - PubMed
-
- Barinaga M. Science. 1994;266:973. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
