

Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome
- PMID: 17914026
- PMCID: PMC2200836
- DOI: 10.1182/blood-2007-04-084533
Membrane cofactor protein mutations in atypical hemolytic uremic syndrome (aHUS), fatal Stx-HUS, C3 glomerulonephritis, and the HELLP syndrome
Abstract
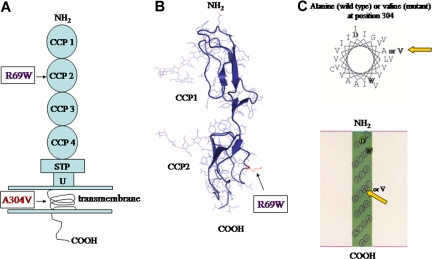
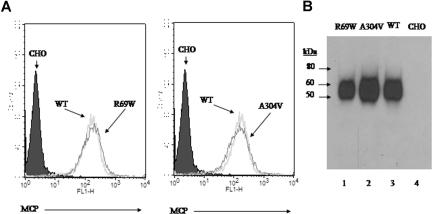
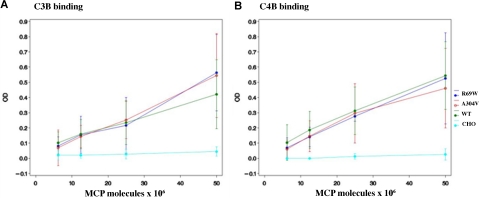
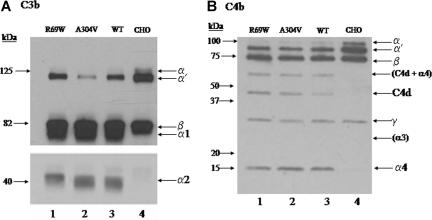
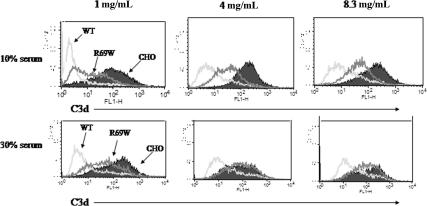
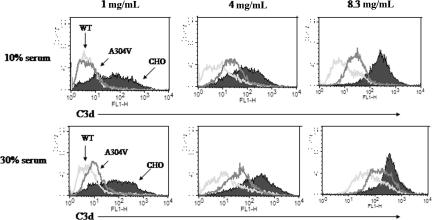
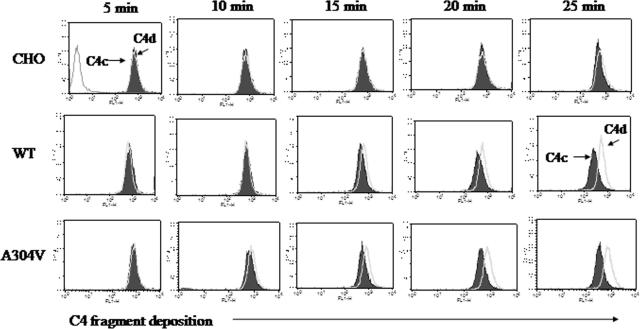
The hemolytic uremic syndrome (HUS) is a triad of microangiopathic hemolytic anemia, thrombocytopenia, and renal impairment. Genetic studies demonstrate that heterozygous mutations of membrane cofactor protein (MCP;CD46) predispose to atypical HUS (aHUS), which is not associated with exposure to Shiga toxin (Stx). Among the initial 25 MCP mutations in patients with aHUS were 2, R69W and A304V, that were expressed normally and for which no dysfunction was found. The R69W mutation is in complement control protein module 2, while A304V is in the hydrophobic transmembrane domain. In addition to 3 patients with aHUS, the A304V mutation was identified in 1 patient each with fatal Stx-HUS, the HELLP (hemolysis, elevated liver enzymes, and low platelets) syndrome, and glomerulonephritis with C3 deposits. A major goal was to assess if these putative mutations lead to defective complement regulation. Permanent cell lines expressing the mutated proteins were complement "challenged," and membrane control of C3 fragment deposition was monitored. Both the R69W and A304V MCP mutations were deficient in their ability to control the alternative pathway of complement activation on a cell surface, illustrating the importance of modeling transmembrane proteins in situ.
Figures
References
-
- Moake JL. Thrombotic microangiopathies. N Engl J Med. 2002;347:589–600. - PubMed
-
- Warwicker P, Goodship THJ, Donne RL, et al. Genetic studies into inherited and sporadic hemolytic uremic syndrome. Kidney Int. 1998;53:836–844. - PubMed
-
- Caprioli J, Bettinaglio P, Zipfel PF, et al. The molecular basis of familial hemolytic uremic syndrome: mutation analysis of factor H gene reveals a hot spot in short consensus repeat 20. J Am Soc Nephrol. 2001;12:297–307. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
