

Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans
- PMID: 17914900
- PMCID: PMC1994989
- DOI: 10.1371/journal.pbio.0050259
Relationship between mitochondrial electron transport chain dysfunction, development, and life extension in Caenorhabditis elegans
Erratum in
- PLoS Biol. 2008 Jan;6(1):e23
- PLoS Biol. 2008 Mar;6(3):e78
- PLoS Biol. 2008 May;6(5):e136
Abstract
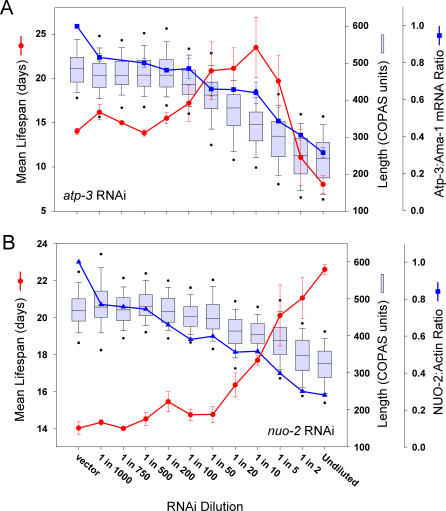
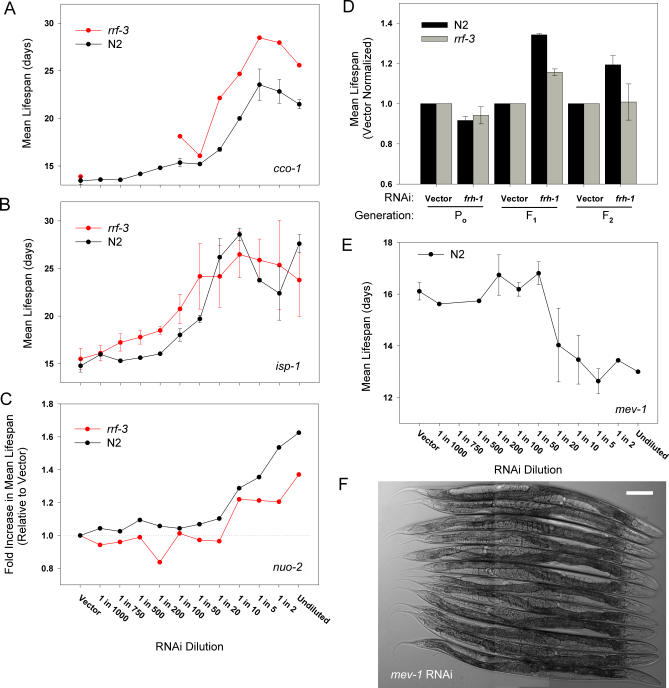
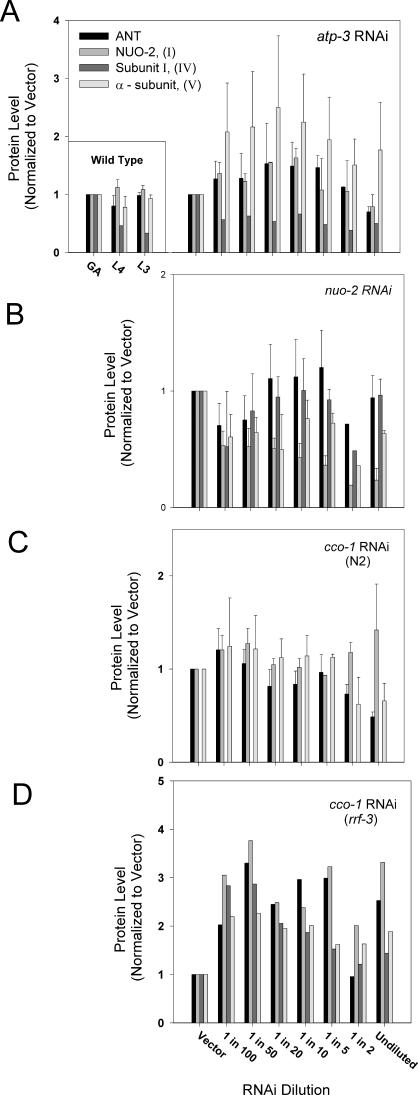
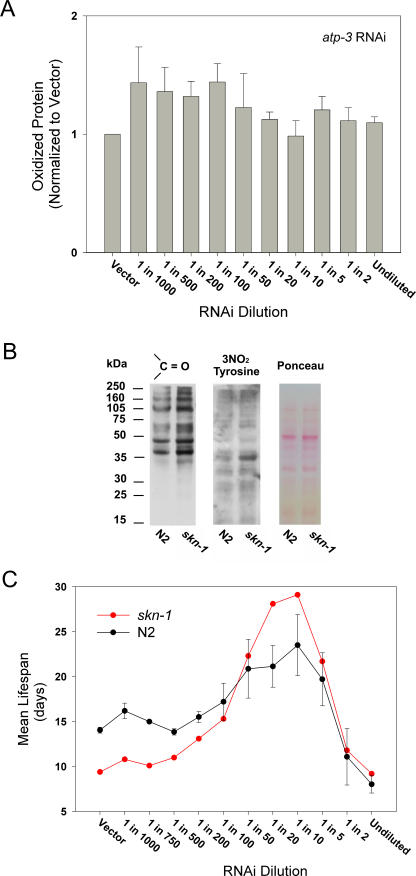
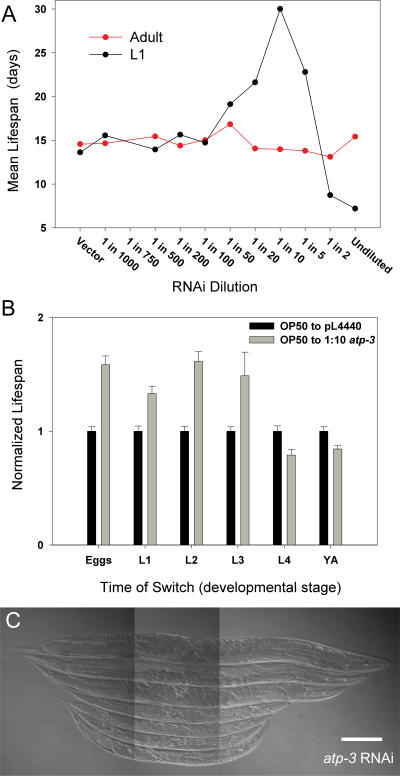
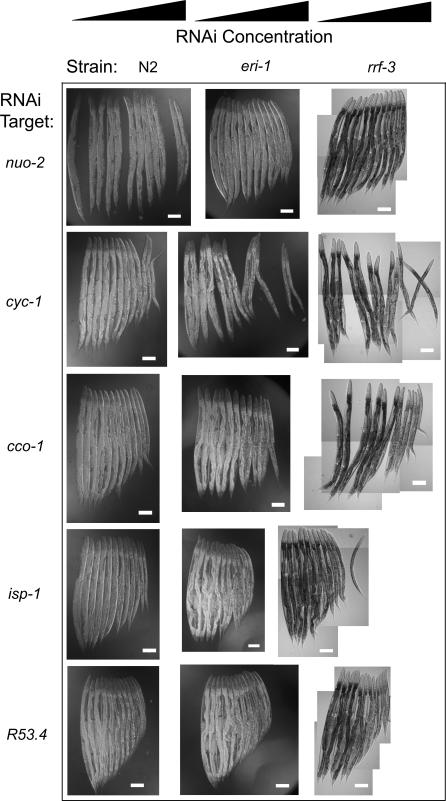
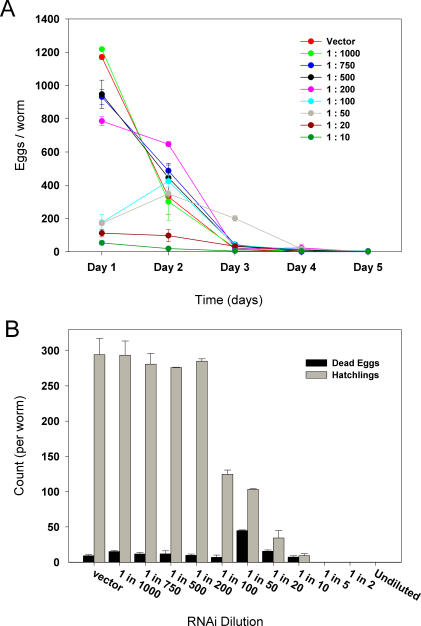
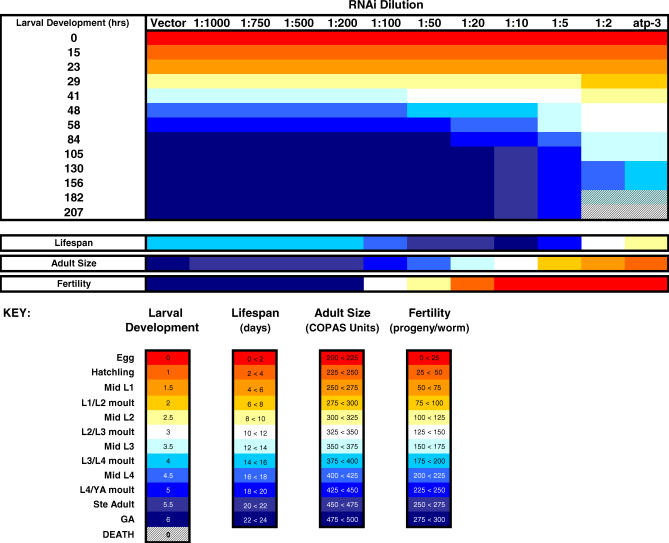
Prior studies have shown that disruption of mitochondrial electron transport chain (ETC) function in the nematode Caenorhabditis elegans can result in life extension. Counter to these findings, many mutations that disrupt ETC function in humans are known to be pathologically life-shortening. In this study, we have undertaken the first formal investigation of the role of partial mitochondrial ETC inhibition and its contribution to the life-extension phenotype of C. elegans. We have developed a novel RNA interference (RNAi) dilution strategy to incrementally reduce the expression level of five genes encoding mitochondrial proteins in C. elegans: atp-3, nuo-2, isp-1, cco-1, and frataxin (frh-1). We observed that each RNAi treatment led to marked alterations in multiple ETC components. Using this dilution technique, we observed a consistent, three-phase lifespan response to increasingly greater inhibition by RNAi: at low levels of inhibition, there was no response, then as inhibition increased, lifespan responded by monotonically lengthening. Finally, at the highest levels of RNAi inhibition, lifespan began to shorten. Indirect measurements of whole-animal oxidative stress showed no correlation with life extension. Instead, larval development, fertility, and adult size all became coordinately affected at the same point at which lifespan began to increase. We show that a specific signal, initiated during the L3/L4 larval stage of development, is sufficient for initiating mitochondrial dysfunction-dependent life extension in C. elegans. This stage of development is characterized by the last somatic cell divisions normally undertaken by C. elegans and also by massive mitochondrial DNA expansion. The coordinate effects of mitochondrial dysfunction on several cell cycle-dependent phenotypes, coupled with recent findings directly linking cell cycle progression with mitochondrial activity in C. elegans, lead us to propose that cell cycle checkpoint control plays a key role in specifying longevity of mitochondrial mutants.
Conflict of interest statement
Figures

Comment in
-
Mitochondria and aging: dilution is the solution.Cell Metab. 2007 Dec;6(6):427-9. doi: 10.1016/j.cmet.2007.11.008. Cell Metab. 2007. PMID: 18054312
References
-
- Campuzano V, Montermini L, Molto MD, Pianese L, Cossee M, et al. Friedreich's ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science. 1996;271:1423–1427. - PubMed
-
- Gerber J, Lill R. Biogenesis of iron-sulfur proteins in eukaryotes: Components, mechanism and pathology. Mitochondrion. 2002;2:71–86. - PubMed
-
- Karlberg T, Schagerlof U, Gakh O, Park S, Ryde U, et al. The structures of frataxin oligomers reveal the mechanism for the delivery and detoxification of iron. Structure. 2006;14:1535–1546. - PubMed
-
- Rea SL. Metabolism in the Caenorhabditis elegans Mit mutants. Exp Gerontol. 2005;40:841–849. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
