

Role of autophagy in the pathogenesis of Pompe disease
- PMID: 17915569
- PMCID: PMC2949326
Role of autophagy in the pathogenesis of Pompe disease
Abstract
In Pompe disease, a deficiency of lysosomal acid alpha-glucosidase, glycogen accumulates in multiple tissues, but clinical manifestations are mainly due to skeletal and cardiac muscle involvement. A major advance has been the development of enzyme replacement therapy (ERT), which recently became available for Pompe patients. Based on clinical and pre-clinical studies, the effective clearance of skeletal muscle glycogen appears to be more difficult than anticipated. Skeletal muscle destruction and resistance to therapy remain unsolved problems. We have found that the cellular pathology in Pompe disease spreads to affect both the endocytic and autophagic pathways, leading to excessive autophagic buildup in therapy resistant muscle fibers of knockout mice. Furthermore, the autophagic buildup had a profound effect on the trafficking and processing of the therapeutic enzyme along the endocytic pathway. These findings may explain why ERT often falls short of reversing the disease process, and point to new avenues for the development of pharmacological intervention.
Figures
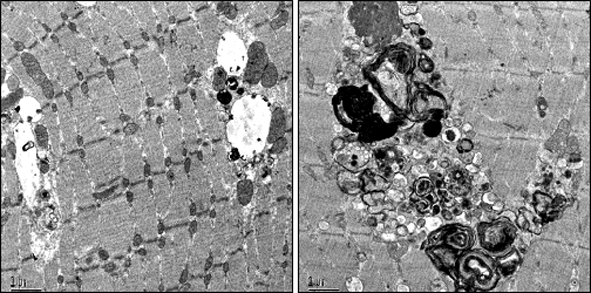

References
-
- Reuser AJ, Van den Hout H, Bijvoet A, et al. Enzyme therapy for Pompe disease: from science to industrial enterprise. Eur J Pediatr 2002;161:106-11. - PubMed
-
- Engel AG, Hirschhorn R, Huie ML. Acid Maltase Deficiency. In: Engel AG, Franzini-Armstrong C, eds. Myology. Third Edition. New York: McGraw-Hill; 2003. p. 1559-86.
-
- Reuser AJ, Kroos MA, Hermans MM, et al. Glycogenosis type II (acid maltase deficiency). Muscle Nerve 1995;3:61-9. - PubMed
-
- Van den Hout JM, Kamphoven JH, Winkel LP, et al. Long-term intravenous treatment of Pompe disease with recombinant human alpha-glucosidase from milk. Pediatrics 2004;113:448-57. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical