

Molecular genetics of late onset glycogen storage disease II in Italy
- PMID: 17915575
- PMCID: PMC2949324
Molecular genetics of late onset glycogen storage disease II in Italy
Abstract
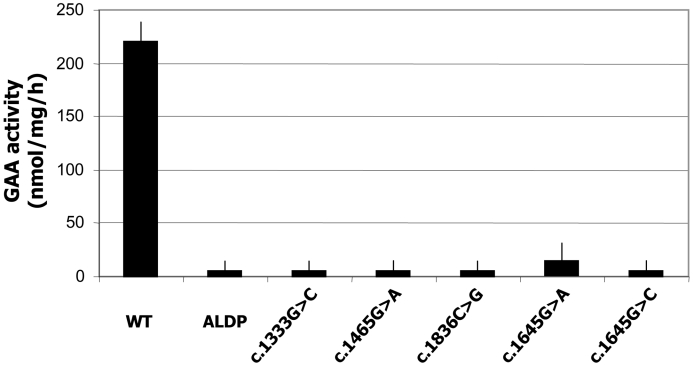
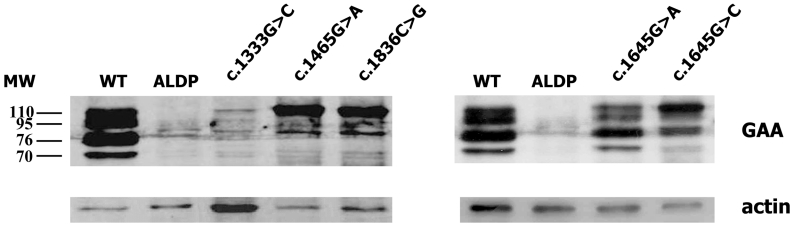
Glycogen Storage Disease Type II (GSDII) is a recessively inherited disorder due to the deficiency of acid alpha-glucosidase (GAA) that results in glycogen accumulation in the lysosomes. The molecular analysis of the GAA gene was performed on 45 Italian patients with late onset GSDII. DHPLC analysis revealed 28 polymorphisms spread all over the GAA gene. Direct sequencing identified the 96% of the mutant alleles, 12 of which are novel. Missense mutations were functionally characterized by enzyme activity and protein processing in a human GAA deficient cell line while splicing mutations were studied by RT-PCR and in silico analysis. A complex allele was also identified carrying three different alterations in cis. All the patients studied carried a severe mutation in combination with a milder one, which explains the late onset of the disease. The c.-32-13T > G was the most frequent mutation, present as compound heterozygote in 85% of the patients as described in other late onset GSDII Caucasian populations. Interestingly, 10 of the 45 patients carried the c.-32-13T > G associated to the severe c.2237G > A (p.W746X) mutation. However, despite the common genotype, patients presented with a wide variability in residual enzyme activity, age of appearance of clinical signs and rate of disease progression, suggesting that other genetic/environment factors may modulate clinical presentation.
Figures
Similar articles
-
Mutation profile of the GAA gene in 40 Italian patients with late onset glycogen storage disease type II.Hum Mutat. 2006 Oct;27(10):999-1006. doi: 10.1002/humu.20374. Hum Mutat. 2006. PMID: 16917947
-
Development of a clinical assay for detection of GAA mutations and characterization of the GAA mutation spectrum in a Canadian cohort of individuals with glycogen storage disease, type II.Mol Genet Metab. 2007 Dec;92(4):325-35. doi: 10.1016/j.ymgme.2007.07.006. Epub 2007 Aug 27. Mol Genet Metab. 2007. PMID: 17723315
-
Molecular diagnosis of German patients with late-onset glycogen storage disease type II.J Inherit Metab Dis. 2008 Dec;31 Suppl 2:S261-5. doi: 10.1007/s10545-008-0820-2. Epub 2008 Jul 10. J Inherit Metab Dis. 2008. PMID: 18607768
-
Extended phenotype description and new molecular findings in late onset glycogen storage disease type II: a northern Italy population study and review of the literature.J Neurol. 2014 Jan;261(1):83-97. doi: 10.1007/s00415-013-7137-2. Epub 2013 Oct 25. J Neurol. 2014. PMID: 24158270 Review.
-
Genetic defects in patients with glycogenosis type II (acid maltase deficiency).Muscle Nerve Suppl. 1995;3:S70-4. doi: 10.1002/mus.880181415. Muscle Nerve Suppl. 1995. PMID: 7603531 Review.
Cited by
-
Distribution of Exonic Variants in Glycogen Synthesis and Catabolism Genes in Late Onset Pompe Disease (LOPD).Curr Issues Mol Biol. 2023 Apr 1;45(4):2847-2860. doi: 10.3390/cimb45040186. Curr Issues Mol Biol. 2023. PMID: 37185710 Free PMC article.
-
Genotype-phenotype correlation in Pompe disease, a step forward.Orphanet J Rare Dis. 2014 Aug 8;9:102. doi: 10.1186/s13023-014-0102-z. Orphanet J Rare Dis. 2014. PMID: 25103075 Free PMC article.
-
Broad variation in phenotypes for common GAA genotypes in Pompe disease.Hum Mutat. 2021 Nov;42(11):1461-1472. doi: 10.1002/humu.24272. Epub 2021 Sep 8. Hum Mutat. 2021. PMID: 34405923 Free PMC article.
-
Clinical and molecular aspects of 30 patients with late-onset Pompe disease (LOPD): unusual features and response to treatment.J Neurol. 2015;262(4):968-78. doi: 10.1007/s00415-015-7664-0. Epub 2015 Feb 12. J Neurol. 2015. PMID: 25673129
-
Genetic counseling in Pompe disease.Acta Myol. 2011 Dec;30(3):179-81. Acta Myol. 2011. PMID: 22616199 Free PMC article.
References
-
- Hirschhorn R, Reuser AJJ. Glycogen storage disease type II: acid α-glucosidase (acid maltase) deficiency, In: Scriver CR, Beaudet AL, Sly WS, Valle D, eds. The metabolic and molecular basis of inherited disease. Vol 3, 8th edn. New York: McGraw-Hill; 2001. p. 3389-420.
-
- Raben N, Plotz P, Byrne BJ. Acid alpha-glucosidase deficiency (glycogenosis type II, Pompe disease). Curr Mol Med 2002;2:145-66. - PubMed
-
- Van den Hout HM, Hop W, van Diggelen OP. The natural course of infantile Pompe’s disease: 20 original cases compared with 133 cases from the literature. Pediatrics 2003;112:332-40. - PubMed
-
- Kishnani PS, Howell RR. Pompe disease in infants and children. J Pediatr 2004;144:35-43. - PubMed
-
- Slonim AE, Bulone L, Ritz S, Goldberg T, Chen A, Martiniuk F. Identification of two subtypes of infantile acid maltase deficiency. J Pediatr 2000;137:283-5. - PubMed
MeSH terms
Substances
Associated data
- Actions
- Actions
LinkOut - more resources
Full Text Sources
Miscellaneous