

Genetic diversity of the obligate intracellular bacterium Chlamydophila pneumoniae by genome-wide analysis of single nucleotide polymorphisms: evidence for highly clonal population structure
- PMID: 17916241
- PMCID: PMC2092436
- DOI: 10.1186/1471-2164-8-355
Genetic diversity of the obligate intracellular bacterium Chlamydophila pneumoniae by genome-wide analysis of single nucleotide polymorphisms: evidence for highly clonal population structure
Abstract
Background: Chlamydophila pneumoniae is an obligate intracellular bacterium that replicates in a biphasic life cycle within eukaryotic host cells. Four published genomes revealed an identity of > 99 %. This remarkable finding raised questions about the existence of distinguishable genotypes in correlation with geographical and anatomical origin.
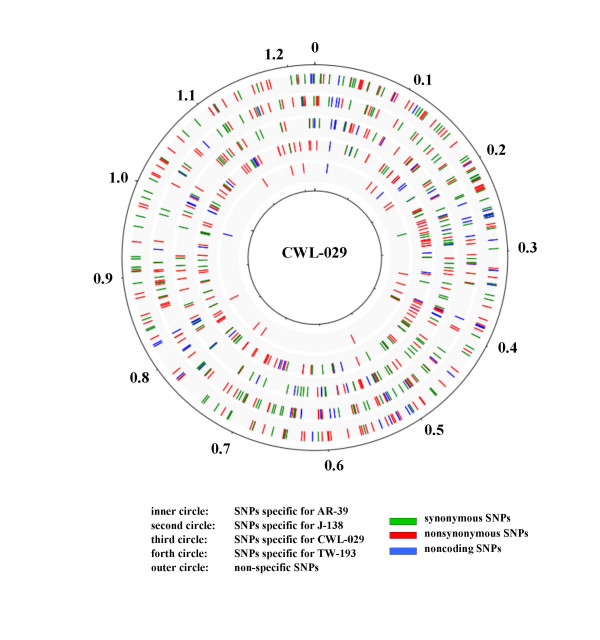
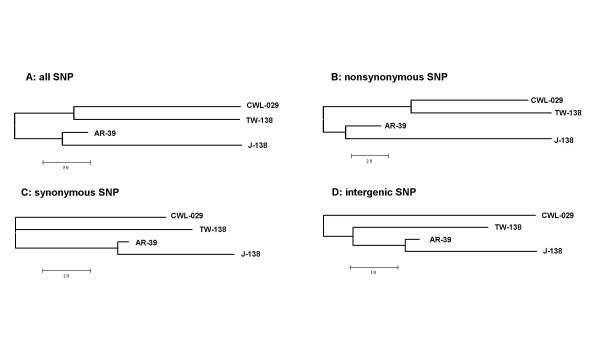
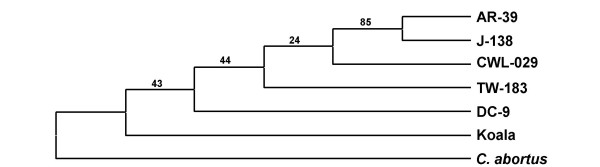
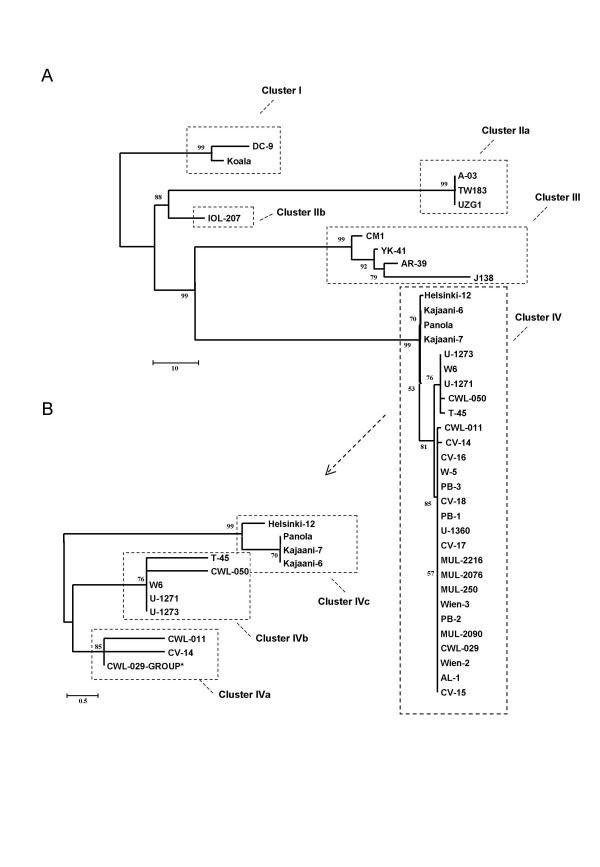
Results: We studied the genetic diversity of C. pneumoniae by analysing synonymous single nucleotide polymorphisms (sSNPs) that are under reduced selection pressure. We conducted an in silico analysis of the four sequenced genomes, chose 232 representative sSNPs and analysed the loci in 38 C. pneumoniae isolates. We identified 15 different genotypes that were separated in four major clusters. Clusters were not associated with anatomical or geographical origin. However, animal lineages are basal on the C. pneumomiae phylogeny, suggesting a recent transmission to humans through successive bottlenecks some 150,000 years ago. A lack of detectable variation in 17 isolates emphasizes the extraordinary genetic conservation of this species and the high clonality of the population. Moreover, the largest cluster, which encompasses 80% of all analysed strains, is an extremely young clade, that went through an important population expansion some 3,300 years ago.
Conclusion: sSNPs have proven useful as a sensitive marker to gain new insights into genetic diversity, population structure and evolutionary history of C. pneumoniae.
Figures
Similar articles
-
Phylogenetic analysis of human Chlamydia pneumoniae strains reveals a distinct Australian indigenous clade that predates European exploration of the continent.BMC Genomics. 2015 Dec 22;16:1094. doi: 10.1186/s12864-015-2281-y. BMC Genomics. 2015. PMID: 26694618 Free PMC article.
-
Multi locus sequence typing of Chlamydiales: clonal groupings within the obligate intracellular bacteria Chlamydia trachomatis.BMC Microbiol. 2008 Feb 28;8:42. doi: 10.1186/1471-2180-8-42. BMC Microbiol. 2008. PMID: 18307777 Free PMC article.
-
Comparative genomic analysis of human Chlamydia pneumoniae isolates from respiratory, brain and cardiac tissues.Genomics. 2015 Dec;106(6):373-83. doi: 10.1016/j.ygeno.2015.09.008. Epub 2015 Sep 28. Genomics. 2015. PMID: 26420648
-
Chlamydia pneumoniae: modern insights into an ancient pathogen.Trends Microbiol. 2013 Mar;21(3):120-8. doi: 10.1016/j.tim.2012.10.009. Epub 2012 Dec 5. Trends Microbiol. 2013. PMID: 23218799 Review.
-
Evolution to a chronic disease niche correlates with increased sensitivity to tryptophan availability for the obligate intracellular bacterium Chlamydia pneumoniae.J Bacteriol. 2014 Jun;196(11):1915-24. doi: 10.1128/JB.01476-14. Epub 2014 Mar 28. J Bacteriol. 2014. PMID: 24682324 Free PMC article. Review.
Cited by
-
Evidence that human Chlamydia pneumoniae was zoonotically acquired.J Bacteriol. 2009 Dec;191(23):7225-33. doi: 10.1128/JB.00746-09. Epub 2009 Sep 11. J Bacteriol. 2009. PMID: 19749045 Free PMC article.
-
The Genetic Transformation of Chlamydia pneumoniae.mSphere. 2018 Oct 10;3(5):e00412-18. doi: 10.1128/mSphere.00412-18. mSphere. 2018. PMID: 30305318 Free PMC article.
-
A molecular survey of Chlamydia spp. infection in commercial poultry and detection of Chlamydia pneumoniae in a commercial turkey flock in Iran.Vet Med Sci. 2023 Sep;9(5):2168-2175. doi: 10.1002/vms3.1244. Epub 2023 Aug 21. Vet Med Sci. 2023. PMID: 37602896 Free PMC article.
-
Chlamydia pneumoniae is genetically diverse in animals and appears to have crossed the host barrier to humans on (at least) two occasions.PLoS Pathog. 2010 May 20;6(5):e1000903. doi: 10.1371/journal.ppat.1000903. PLoS Pathog. 2010. PMID: 20502684 Free PMC article.
-
Diversification and Distribution of Ruminant Chlamydia abortus Clones Assessed by MLST and MLVA.PLoS One. 2015 May 22;10(5):e0126433. doi: 10.1371/journal.pone.0126433. eCollection 2015. PLoS One. 2015. PMID: 26001070 Free PMC article.
References
-
- Stephens RS. Chlamydial Evolution: A billion years and counting. In: Schachter, J, editor. Chlamydial Infections Prodeedings of the Tenth International Symposium on Human Chlamydial Infections. Antalya, Turkey; 2002.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
