

qPIPSA: relating enzymatic kinetic parameters and interaction fields
- PMID: 17919319
- PMCID: PMC2174957
- DOI: 10.1186/1471-2105-8-373
qPIPSA: relating enzymatic kinetic parameters and interaction fields
Abstract
Background: The simulation of metabolic networks in quantitative systems biology requires the assignment of enzymatic kinetic parameters. Experimentally determined values are often not available and therefore computational methods to estimate these parameters are needed. It is possible to use the three-dimensional structure of an enzyme to perform simulations of a reaction and derive kinetic parameters. However, this is computationally demanding and requires detailed knowledge of the enzyme mechanism. We have therefore sought to develop a general, simple and computationally efficient procedure to relate protein structural information to enzymatic kinetic parameters that allows consistency between the kinetic and structural information to be checked and estimation of kinetic constants for structurally and mechanistically similar enzymes.
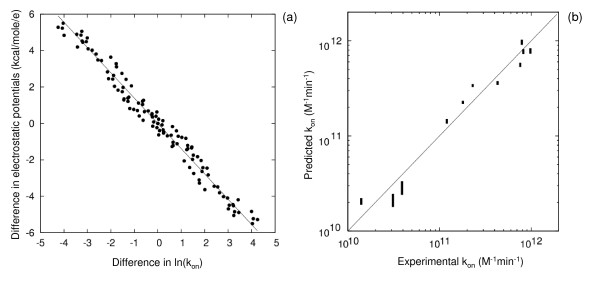
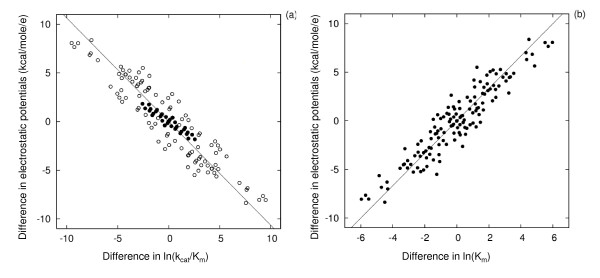
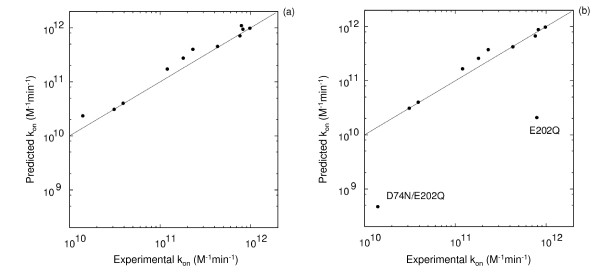
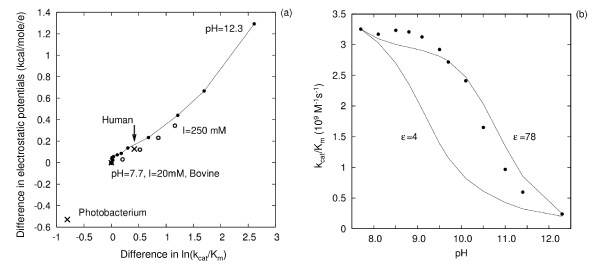
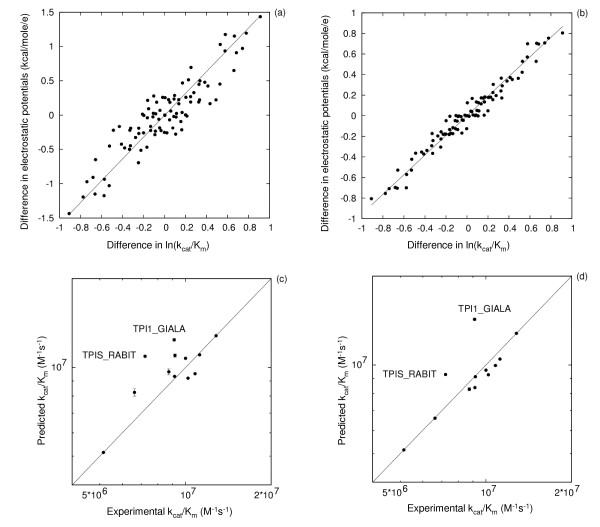
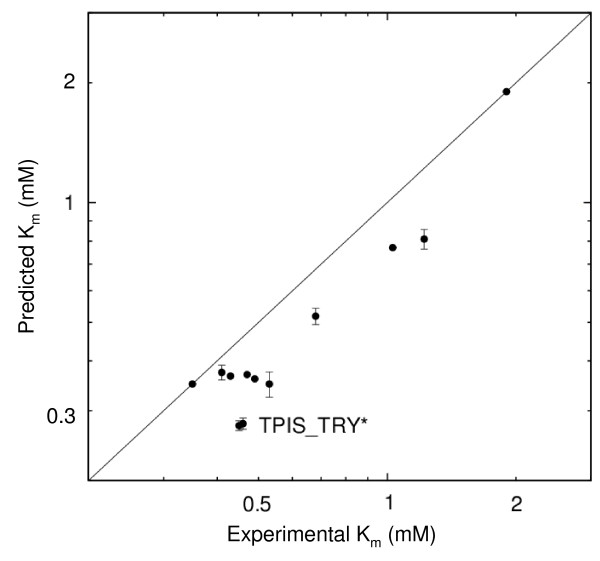
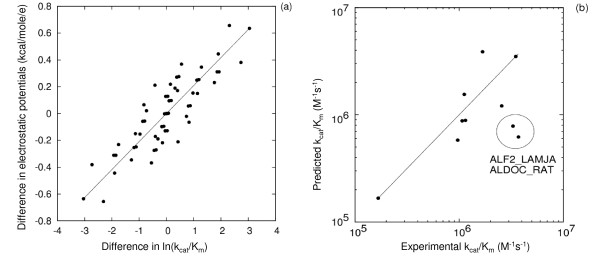
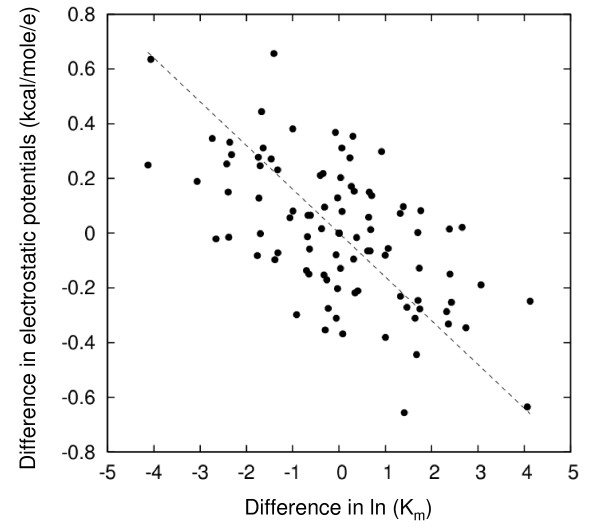
Results: We describe qPIPSA: quantitative Protein Interaction Property Similarity Analysis. In this analysis, molecular interaction fields, for example, electrostatic potentials, are computed from the enzyme structures. Differences in molecular interaction fields between enzymes are then related to the ratios of their kinetic parameters. This procedure can be used to estimate unknown kinetic parameters when enzyme structural information is available and kinetic parameters have been measured for related enzymes or were obtained under different conditions. The detailed interaction of the enzyme with substrate or cofactors is not modeled and is assumed to be similar for all the proteins compared. The protein structure modeling protocol employed ensures that differences between models reflect genuine differences between the protein sequences, rather than random fluctuations in protein structure.
Conclusion: Provided that the experimental conditions and the protein structural models refer to the same protein state or conformation, correlations between interaction fields and kinetic parameters can be established for sets of related enzymes. Outliers may arise due to variation in the importance of different contributions to the kinetic parameters, such as protein stability and conformational changes. The qPIPSA approach can assist in the validation as well as estimation of kinetic parameters, and provide insights into enzyme mechanism.
Figures
Similar articles
-
Calculating enzyme kinetic parameters from protein structures.Biochem Soc Trans. 2008 Feb;36(Pt 1):51-4. doi: 10.1042/BST0360051. Biochem Soc Trans. 2008. PMID: 18208384 Review.
-
webPIPSA: a web server for the comparison of protein interaction properties.Nucleic Acids Res. 2008 Jul 1;36(Web Server issue):W276-80. doi: 10.1093/nar/gkn181. Epub 2008 Apr 17. Nucleic Acids Res. 2008. PMID: 18420653 Free PMC article.
-
Manipulating single enzymes by an external harmonic force.Phys Rev Lett. 2007 Apr 20;98(16):168302. doi: 10.1103/PhysRevLett.98.168302. Epub 2007 Apr 19. Phys Rev Lett. 2007. PMID: 17501470
-
A novel method for determining kinetic parameters of dissociating enzyme systems.Anal Biochem. 1998 Nov 1;264(1):8-21. doi: 10.1006/abio.1998.2818. Anal Biochem. 1998. PMID: 9784182
-
A century of enzyme kinetic analysis, 1913 to 2013.FEBS Lett. 2013 Sep 2;587(17):2753-66. doi: 10.1016/j.febslet.2013.07.012. Epub 2013 Jul 12. FEBS Lett. 2013. PMID: 23850893 Free PMC article. Review.
Cited by
-
Web-based applications for building, managing and analysing kinetic models of biological systems.Brief Bioinform. 2009 Jan;10(1):65-74. doi: 10.1093/bib/bbn039. Epub 2008 Sep 19. Brief Bioinform. 2009. PMID: 18805901 Free PMC article.
-
NNKcat: deep neural network to predict catalytic constants (Kcat) by integrating protein sequence and substrate structure with enhanced data imbalance handling.Brief Bioinform. 2025 May 1;26(3):bbaf212. doi: 10.1093/bib/bbaf212. Brief Bioinform. 2025. PMID: 40370097 Free PMC article.
-
Accuracy of functional surfaces on comparatively modeled protein structures.J Struct Funct Genomics. 2011 Jul;12(2):97-107. doi: 10.1007/s10969-011-9109-z. Epub 2011 May 4. J Struct Funct Genomics. 2011. PMID: 21541664 Free PMC article.
-
Computational study of the human dystrophin repeats: interaction properties and molecular dynamics.PLoS One. 2011;6(8):e23819. doi: 10.1371/journal.pone.0023819. Epub 2011 Aug 25. PLoS One. 2011. PMID: 21901138 Free PMC article.
-
Providing data science support for systems pharmacology and its implications to drug discovery.Expert Opin Drug Discov. 2016;11(3):241-56. doi: 10.1517/17460441.2016.1135126. Epub 2016 Jan 9. Expert Opin Drug Discov. 2016. PMID: 26689499 Free PMC article. Review.
References
-
- Kettner C, Hicks MG. Chaos in the world of enzymes - How valid is functional characterization without methodological experimental data? In: Hicks MG and Kettner C, Logos Verlag; Berlin, editor. Experimental standard conditions of enzyme characterization, Proceedings of the 1st International Beilstein Symposium. Ruedesheim/Rhein; 2003. pp. 1–16.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
