

Subhepatotoxic exposure to arsenic enhances lipopolysaccharide-induced liver injury in mice
- PMID: 17919673
- PMCID: PMC2246049
- DOI: 10.1016/j.taap.2007.08.020
Subhepatotoxic exposure to arsenic enhances lipopolysaccharide-induced liver injury in mice
Abstract
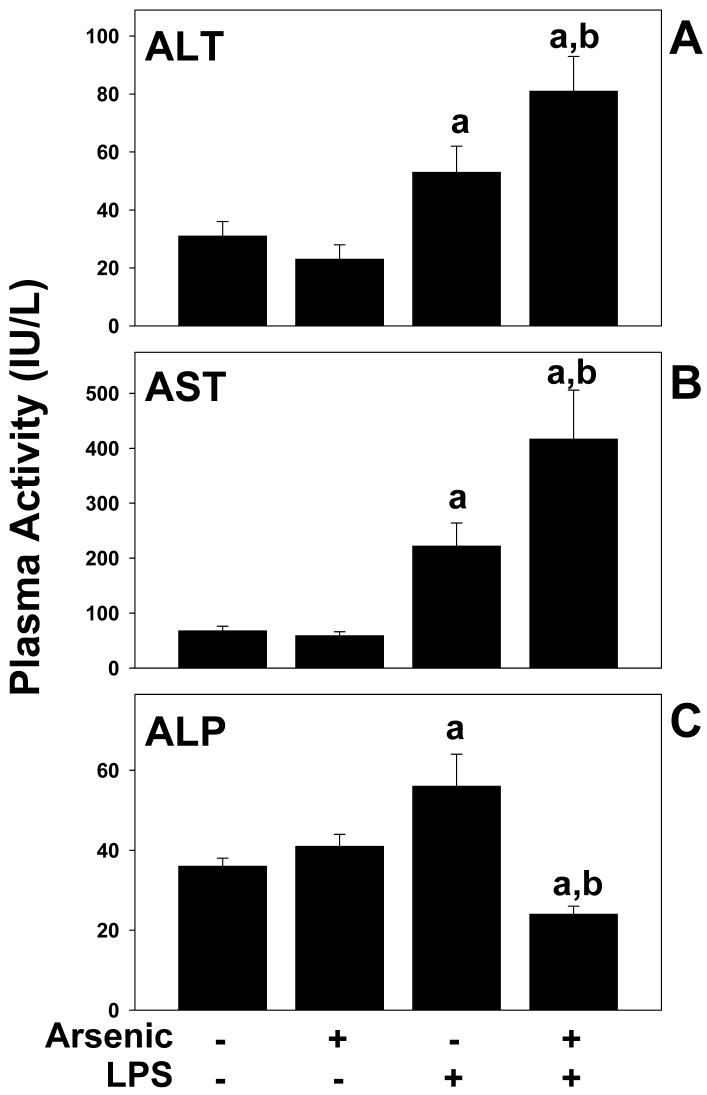
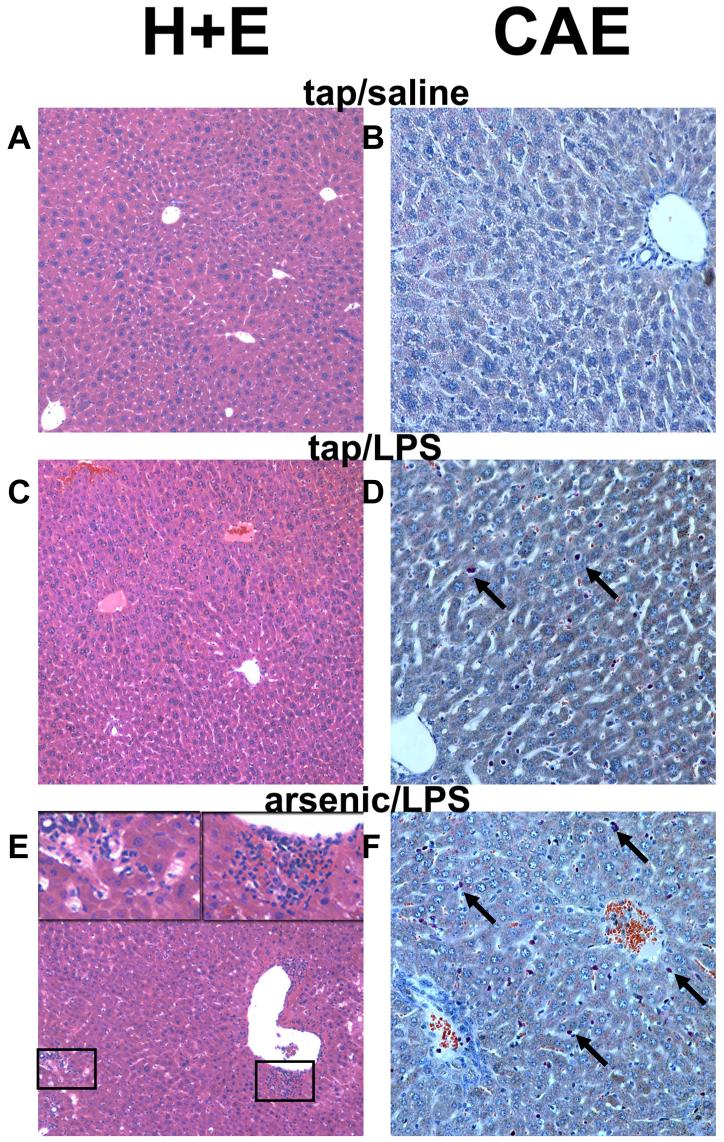
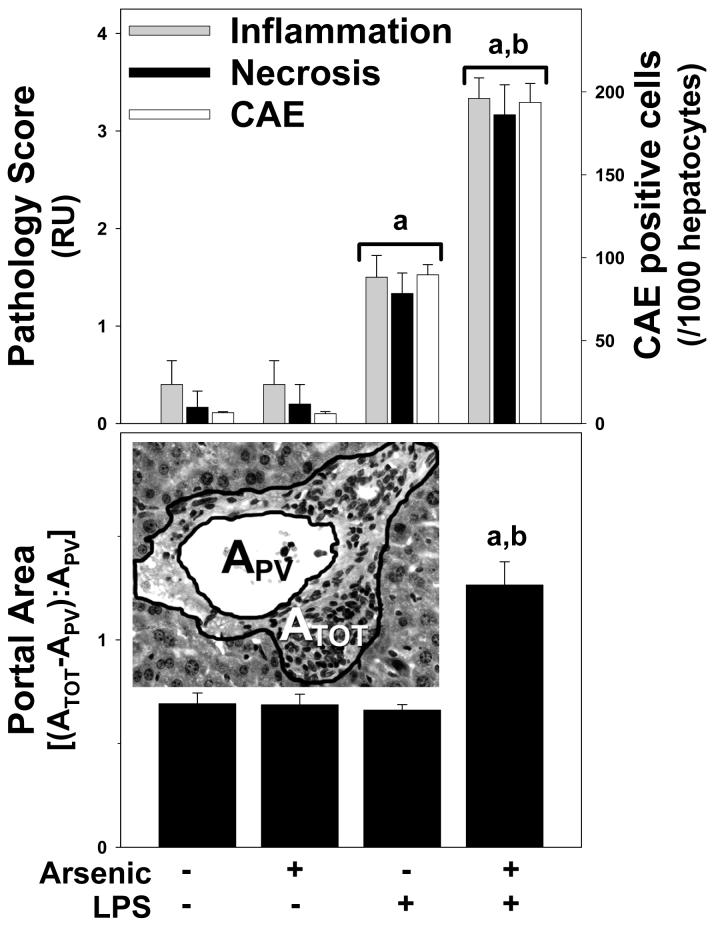
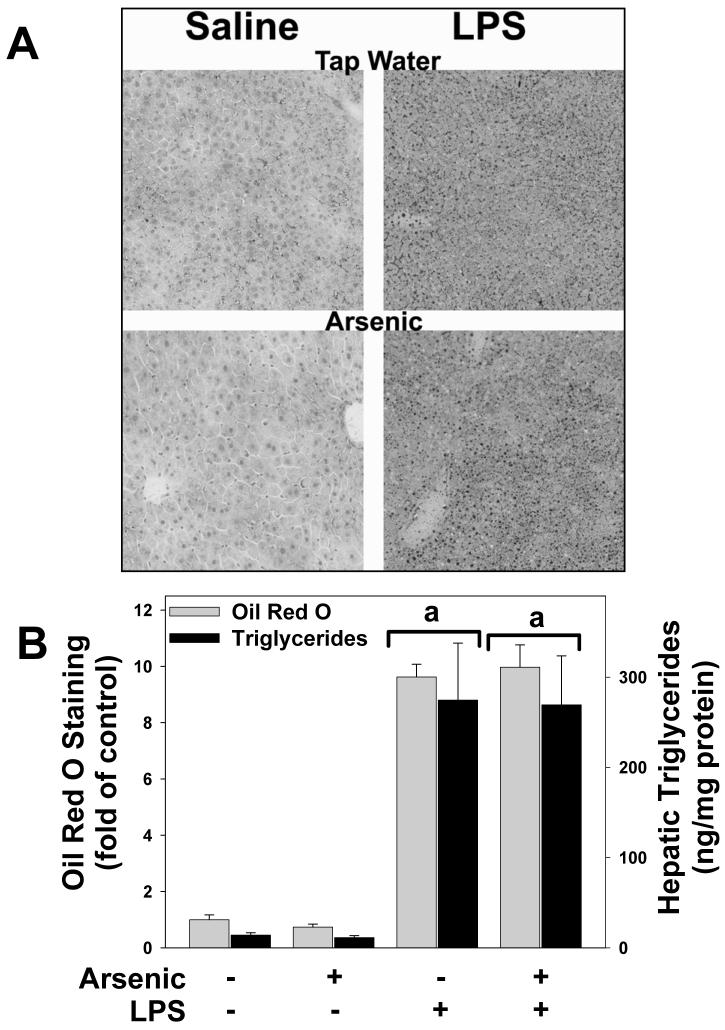
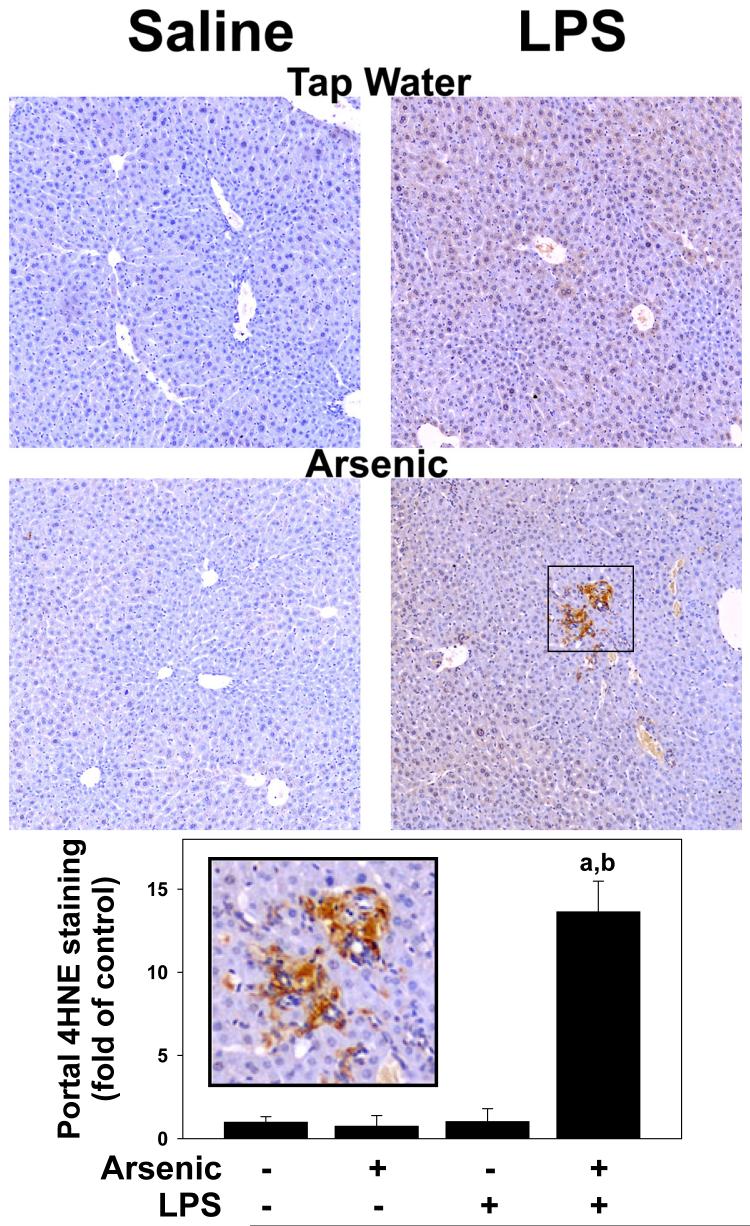
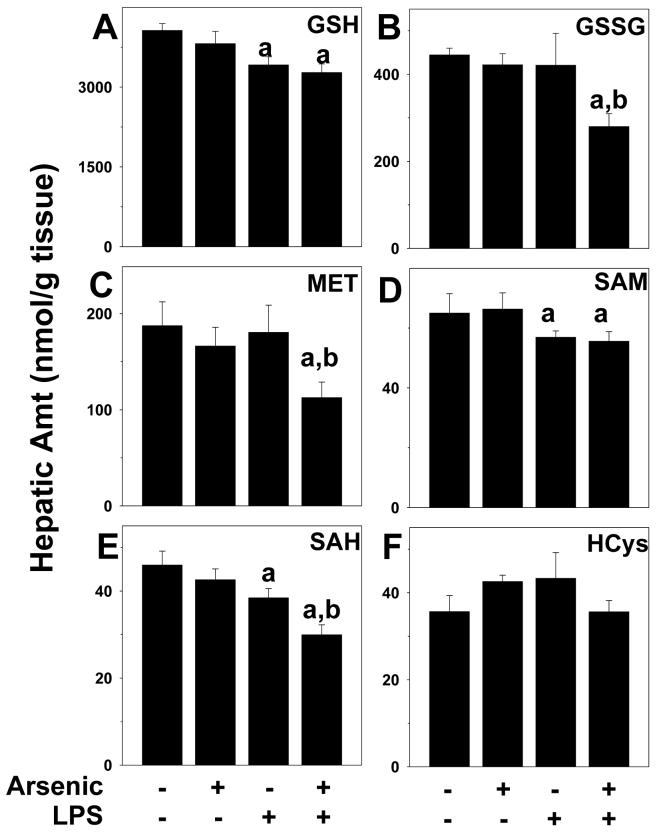
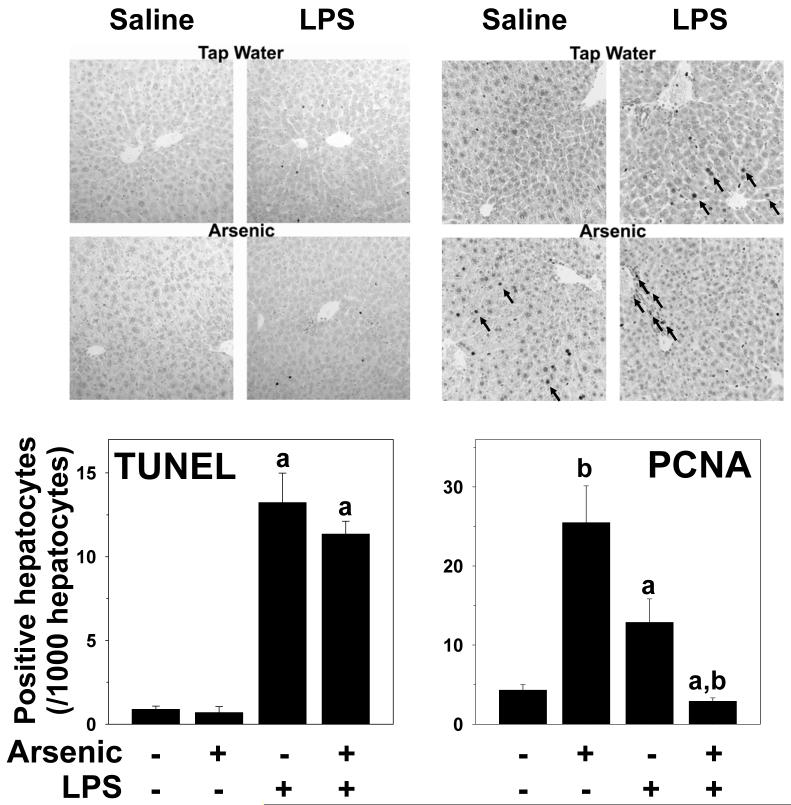
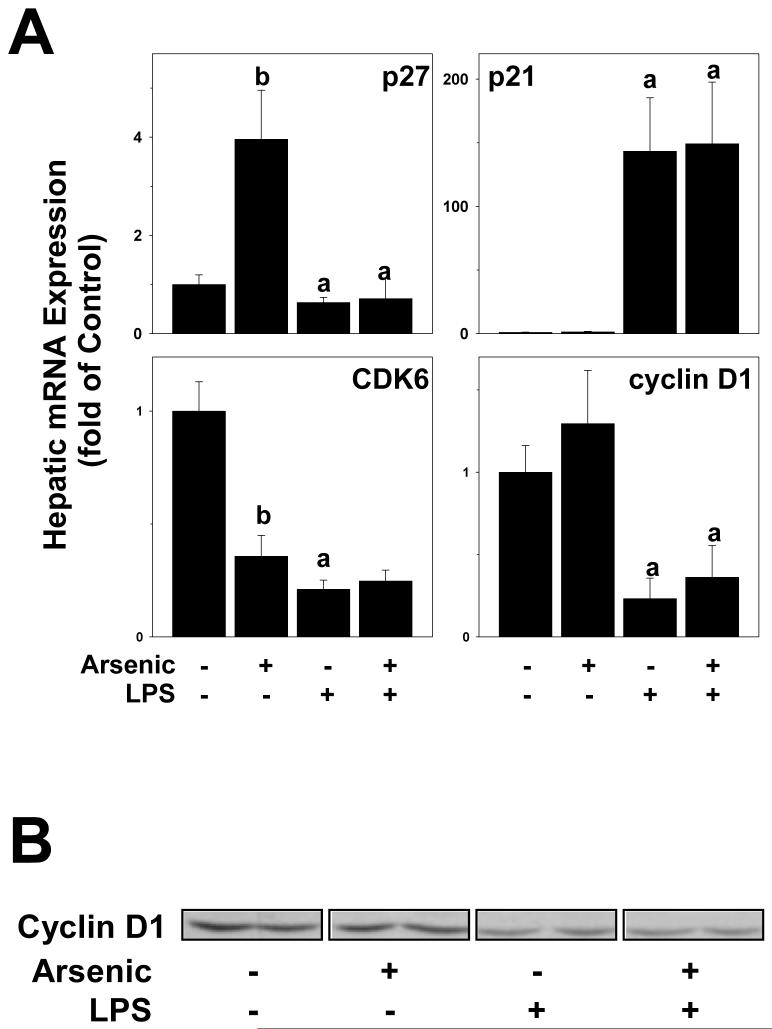
Exposure to arsenic via drinking water is a serious health concern in the US. Whereas studies have identified arsenic alone as an independent risk factor for liver disease, concentrations of arsenic required to damage this organ are generally higher than found in the US water supply. The purpose of the current study was to test the hypothesis that arsenic (at subhepatotoxic doses) may also sensitize the liver to a second hepatotoxin. To test this hypothesis, the effect of chronic exposure to arsenic on liver damage caused by acute lipopolysaccharide (LPS) was determined in mice. Male C57Bl/6J mice (4-6 weeks) were exposed to arsenic (49 ppm as sodium arsenite in drinking water). After 7 months of exposure, animals were injected with LPS (10 mg/kg i.p.) and sacrificed 24 h later. Arsenic alone caused no overt hepatotoxicity, as determined by plasma enzymes and histology. In contrast, arsenic exposure dramatically enhanced liver damage caused by LPS, increasing the number and size of necroinflammatory foci. This effect of arsenic was coupled with increases in indices of oxidative stress (4-HNE adducts, depletion of GSH and methionine pools). The number of apoptotic (TUNEL) hepatocytes was similar in the LPS and arsenic/LPS groups. In contrast, arsenic pre-exposure blunted the increase in proliferating (PCNA) hepatocytes caused by LPS; this change in the balance between cell death and proliferation was coupled with a robust loss of liver weight in the arsenic/LPS compared to the LPS alone group. The impairment of proliferation after LPS caused by arsenic was also coupled with alterations in the expression of key mediators of cell cycle progression (p27, p21, CDK6 and Cyclin D1). Taken together, these results suggest that arsenic, at doses that are not overtly hepatotoxic per se, significantly enhances LPS-induced liver injury. These results further suggest that arsenic levels in the drinking water may be a risk modifier for the development of chronic liver diseases.
Figures
References
-
- Arteel G, Marsano L, Mendez C, Bentley F, McClain CJ. Advances in alcoholic liver disease. Best. Pract. Res. Clin. Gastroenterol. 2003;17:625–647. - PubMed
-
- Caldon CE, Daly RJ, Sutherland RL, Musgrove EA. Cell cycle control in breast cancer cells. J. Cell Biochem. 2006;97:261–274. - PubMed
-
- Chen H, Li S, Liu J, Diwan BA, Barrett JC, Waalkes MP. Chronic inorganic arsenic exposure induces hepatic global and individual gene hypomethylation: implications for arsenic hepatocarcinogenesis. Carcinogenesis. 2004;25:1779–1786. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
