

Characterization of an immunodeficient mouse model of mucopolysaccharidosis type I suitable for preclinical testing of human stem cell and gene therapy
- PMID: 17920451
- PMCID: PMC2148227
- DOI: 10.1016/j.brainresbull.2007.07.018
Characterization of an immunodeficient mouse model of mucopolysaccharidosis type I suitable for preclinical testing of human stem cell and gene therapy
Erratum in
- Brain Res Bull. 2008 Aug 15;76(6):640-1
Abstract
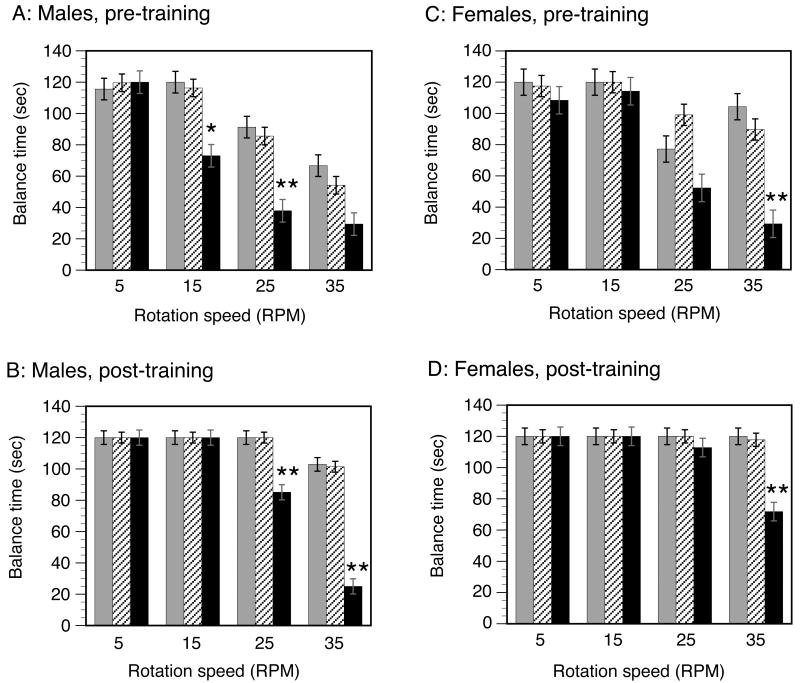
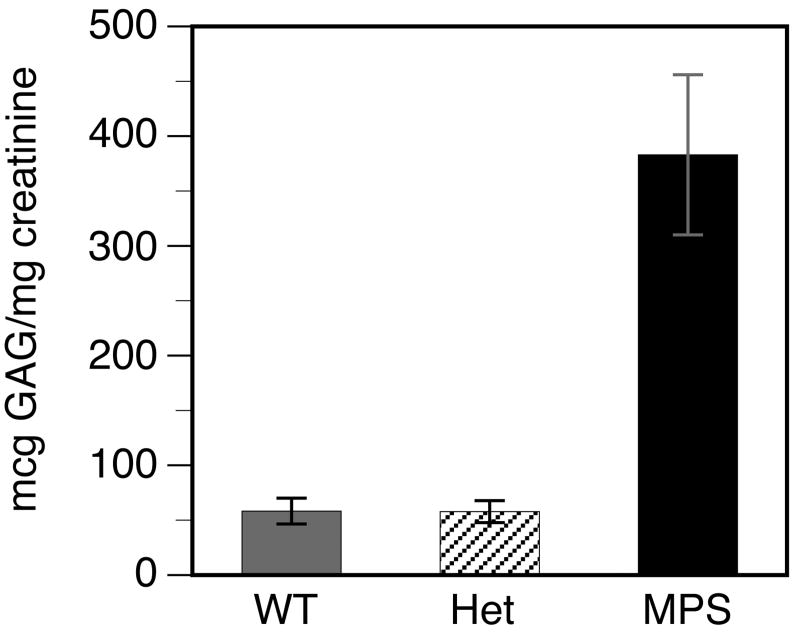
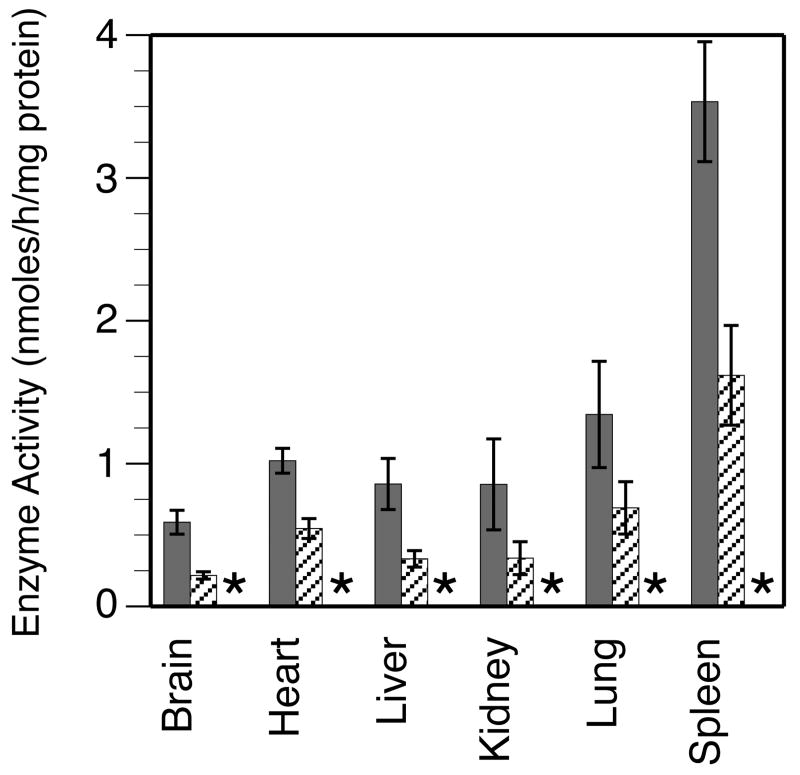
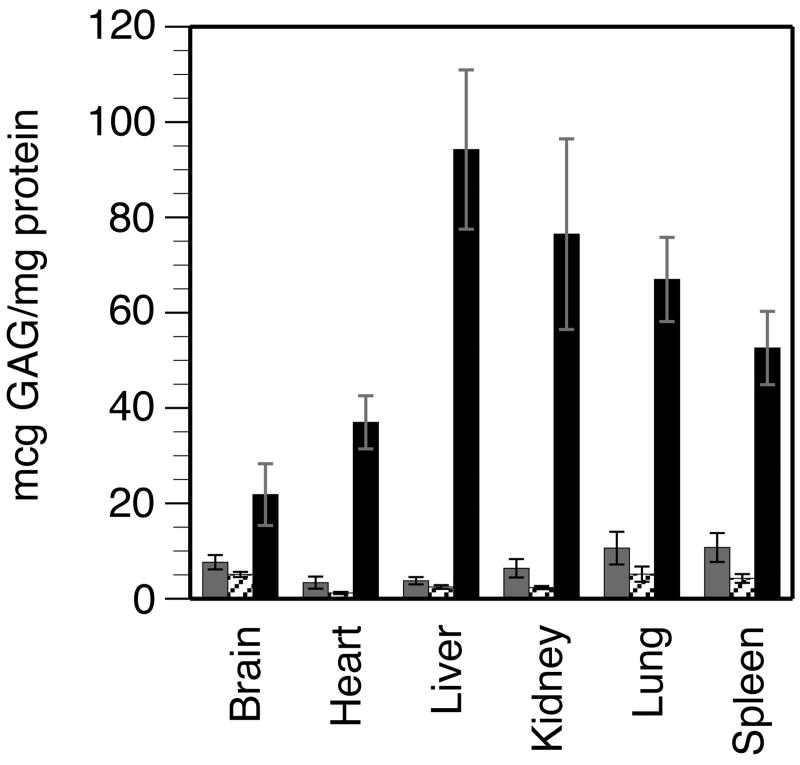
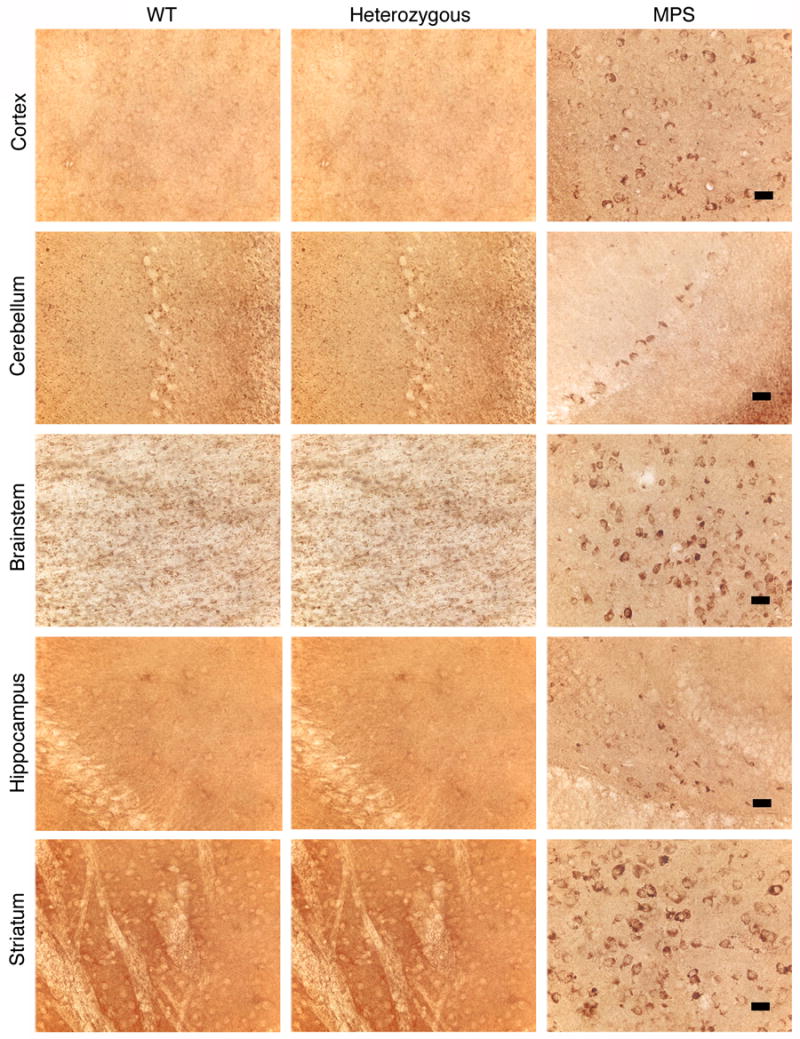
Mucopolysaccharidosis type I (MPS-I or Hurler syndrome) is an inherited deficiency of the lysosomal glycosaminoglycan (GAG)-degrading enzyme alpha-l-iduronidase (IDUA) in which GAG accumulation causes progressive multi-system dysfunction and death. Early allogeneic hematopoietic stem cell transplantation (HSCT) ameliorates clinical features and extends life but is not available to all patients, and inadequately corrects its most devastating features including mental retardation and skeletal deformities. To test novel therapies, we characterized an immunodeficient MPS-I mouse model less likely to develop immune reactions to transplanted human or gene-corrected cells or secreted IDUA. In the liver, spleen, heart, lung, kidney and brain of NOD/SCID/MPS-I mice IDUA was undetectable, and reduced to half in heterozygotes. MPS-I mice developed marked GAG accumulation (3-38-fold) in these organs. Neuropathological examination showed GM(3) ganglioside accumulation in the striatum, cerebral peduncles, cerebellum and ventral brainstem of MPS-I mice. Urinary GAG excretion (6.5-fold higher in MPS-I mice) provided a non-invasive and reliable method suitable for serially following the biochemical efficacy of therapeutic interventions. We identified and validated using rigorous biostatistical methods, a highly reproducible method for evaluating sensorimotor function and motor skills development. This Rotarod test revealed marked abnormalities in sensorimotor integration involving the cerebellum, striatum, proprioceptive pathways, motor cortex, and in acquisition of motor coordination. NOD/SCID/MPS-I mice exhibit many of the clinical, skeletal, pathological and behavioral abnormalities of human MPS-I, and provide an extremely suitable animal model for assessing the systemic and neurological effects of human stem cell transplantation and gene therapeutic approaches, using the above techniques to measure efficacy.
Conflict of interest statement
Figures
Similar articles
-
Intracerebroventricular transplantation of human bone marrow-derived multipotent progenitor cells in an immunodeficient mouse model of mucopolysaccharidosis type I (MPS-I).Cell Transplant. 2012;21(7):1577-93. doi: 10.3727/096368912X636894. Cell Transplant. 2012. PMID: 22472595
-
A novel, long-lived, and highly engraftable immunodeficient mouse model of mucopolysaccharidosis type I.Mol Ther Methods Clin Dev. 2015 Feb 11;2:14068. doi: 10.1038/mtm.2014.68. eCollection 2015. Mol Ther Methods Clin Dev. 2015. PMID: 26052536 Free PMC article.
-
Comparison of Endovascular and Intraventricular Gene Therapy With Adeno-Associated Virus-α-L-Iduronidase for Hurler Disease.Neurosurgery. 2014 Jan;74(1):99-111. doi: 10.1227/NEU.0000000000000157. Neurosurgery. 2014. PMID: 24077583 Free PMC article.
-
Mucopolysaccharidosis type I.Pediatr Endocrinol Rev. 2014 Sep;12 Suppl 1:102-6. Pediatr Endocrinol Rev. 2014. PMID: 25345091 Review.
-
[Mucopolysaccharidosis: clinical features, diagnosis and management].Rev Chil Pediatr. 2016 Jul-Aug;87(4):295-304. doi: 10.1016/j.rchipe.2015.10.004. Epub 2015 Nov 21. Rev Chil Pediatr. 2016. PMID: 26613630 Review. Spanish.
Cited by
-
Assessment of dysmyelination with RAFFn MRI: application to murine MPS I.PLoS One. 2015 Feb 13;10(2):e0116788. doi: 10.1371/journal.pone.0116788. eCollection 2015. PLoS One. 2015. PMID: 25680196 Free PMC article.
-
Contribution of the innate and adaptive immune systems to aortic dilation in murine mucopolysaccharidosis type I.Mol Genet Metab. 2022 Mar;135(3):193-205. doi: 10.1016/j.ymgme.2022.01.104. Epub 2022 Feb 3. Mol Genet Metab. 2022. PMID: 35165009 Free PMC article.
-
Quantitative analysis of α-L-iduronidase expression in immunocompetent mice treated with the Sleeping Beauty transposon system.PLoS One. 2013 Oct 21;8(10):e78161. doi: 10.1371/journal.pone.0078161. eCollection 2013. PLoS One. 2013. PMID: 24205141 Free PMC article.
-
AAV Gene Therapy for MPS1-associated Corneal Blindness.Sci Rep. 2016 Feb 22;6:22131. doi: 10.1038/srep22131. Sci Rep. 2016. PMID: 26899286 Free PMC article.
-
Lipid composition of whole brain and cerebellum in Hurler syndrome (MPS IH) mice.Neurochem Res. 2011 Sep;36(9):1669-76. doi: 10.1007/s11064-011-0400-y. Epub 2011 Jan 21. Neurochem Res. 2011. PMID: 21253856
References
-
- Bhatia M, Bonnett D, Murdoch B, Gan OI, Dick JE. A newly discovered class of human hematopoietic cells with SCID-repopulating activity. Nat Med. 1998;4:1038–45. - PubMed
-
- Bradford MM. A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem. 1976;72:248–54. - PubMed
-
- Braunlin E, Mackey-Bojack S, Panoskaltsis-Mortari A, Berry JM, McElmurry RT, Riddle M, Sun LY, Clarke LA, Tolar J, Blazar BR. Cardiac functional and histopathologic findings in humans and mice with mucopolysaccharidosis type I: implications for assessment of therapeutic interventions in hurler syndrome. Pediatr Res. 2006;59:27–32. - PubMed
-
- Chandrasekhar S, Esterman MA, Hoffman HA. Microdetermination of proteoglycans and glycosaminglycans in the presence of guanidine hydrochloride. Anal Biochem. 1987;161:103–8. - PubMed
-
- Clarke LA, Russell CS, Pownall S, Warrington CL, Borowski A, Dimmick JE, Toone J, Jirik FR. Murine mucopolysaccharidosis type I: targeted disruption of the murine alpha-L-iduronidase gene. Hum Mol Genet. 1997;6:503–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
