

An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for membrane fusion and overcomes diverse entry limitations
- PMID: 17920626
- PMCID: PMC2704606
- DOI: 10.1016/j.jmb.2007.09.016
An allosteric rheostat in HIV-1 gp120 reduces CCR5 stoichiometry required for membrane fusion and overcomes diverse entry limitations
Abstract
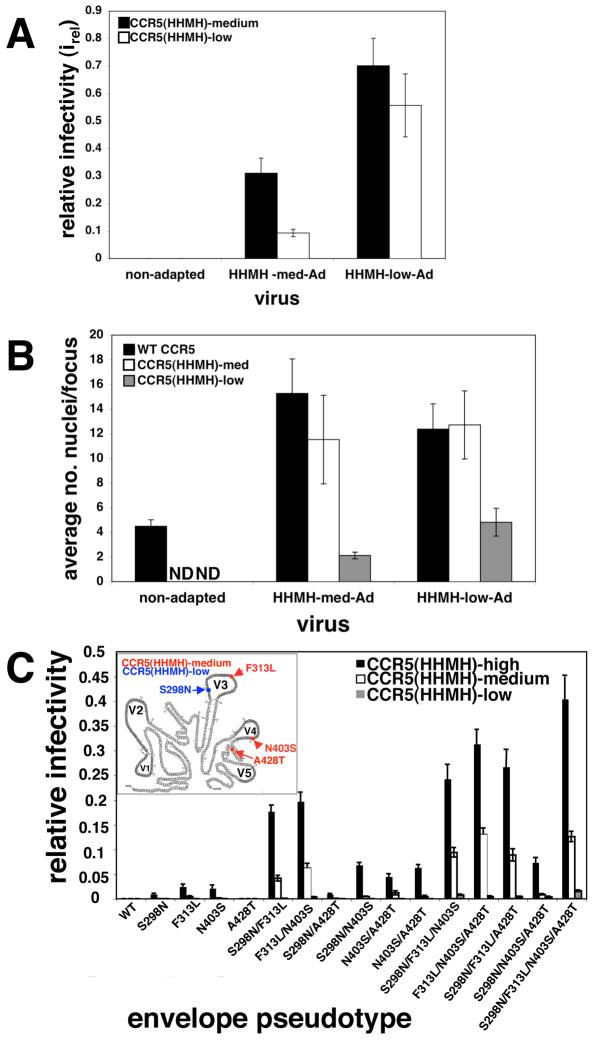
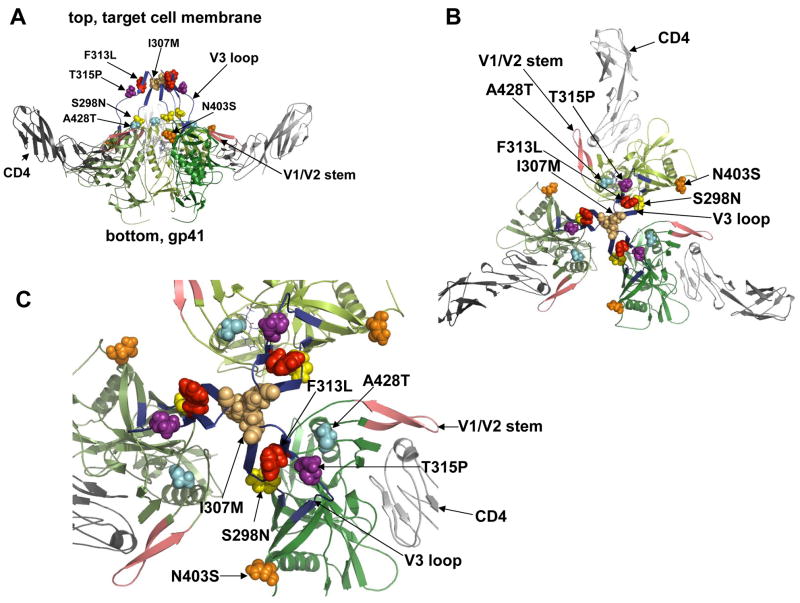
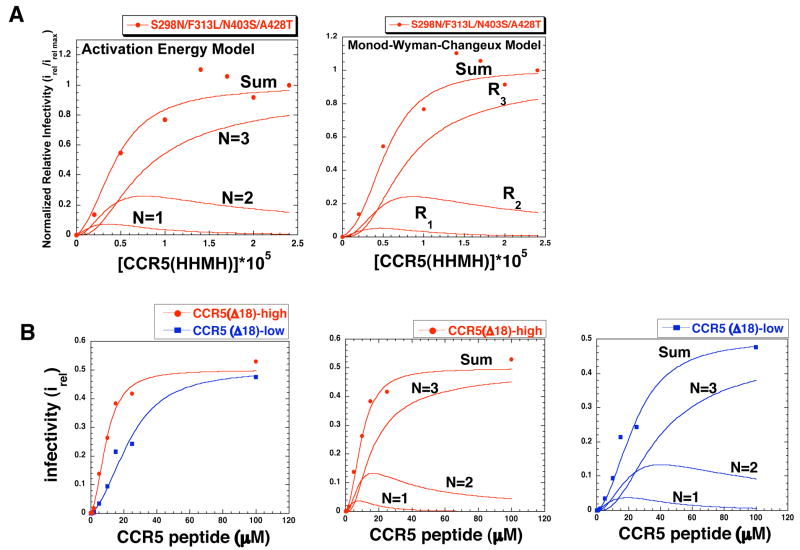
Binding of the human immunodeficiency virus (HIV-1) envelope glycoprotein gp120 to the CCR5 co-receptor reduces constraints on the metastable transmembrane subunit gp41, thereby enabling gp41 refolding, fusion of viral and cellular membranes, and infection. We previously isolated adapted HIV-1(JRCSF) variants that more efficiently use mutant CCR5s, including CCR5(Delta18) lacking the important tyrosine sulfate-containing amino terminus. Effects of mutant CCR5 concentrations on HIV-1 infectivities were highly cooperative, implying that several may be required. However, because wild-type CCR5 efficiently mediates infections at trace concentrations that were difficult to measure accurately, analyses of its cooperativity were not feasible. New HIV-1(JRCSF) variants efficiently use CCR5(HHMH), a chimera containing murine extracellular loop 2. The adapted virus induces large syncytia in cells containing either wild-type or mutant CCR5s and has multiple gp120 mutations that occurred independently in CCR5(Delta18)-adapted virus. Accordingly, these variants interchangeably use CCR5(HHMH) or CCR5(Delta18). Additional analyses strongly support a novel energetic model for allosteric proteins, implying that the adaptive mutations reduce quaternary constraints holding gp41, thus lowering the activation energy barrier for membrane fusion without affecting bonds to specific CCR5 sites. In accordance with this mechanism, highly adapted HIV-1s require only one associated CCR5(HHMH), whereas poorly adapted viruses require several. However, because they are allosteric ensembles, complexes with additional co-receptors fuse more rapidly and efficiently than minimal ones. Similarly, wild-type HIV-1(JRCSF) is highly adapted to wild-type CCR5 and minimally requires one. The adaptive mutations cause resistances to diverse entry inhibitors and cluster appropriately in the gp120 trimer interface overlying gp41. We conclude that membrane fusion complexes are allosteric machines with an ensemble of compositions, and that HIV-1 adapts to entry limitations by gp120 mutations that reduce its allosteric hold on gp41. These results provide an important foundation for understanding the mechanisms that control membrane fusion and HIV-1's facile adaptability.
Figures
Similar articles
-
Adaptive mutations in the V3 loop of gp120 enhance fusogenicity of human immunodeficiency virus type 1 and enable use of a CCR5 coreceptor that lacks the amino-terminal sulfated region.J Virol. 2001 Dec;75(24):12266-78. doi: 10.1128/JVI.75.24.12266-12278.2001. J Virol. 2001. PMID: 11711617 Free PMC article.
-
Variants of human immunodeficiency virus type 1 that efficiently use CCR5 lacking the tyrosine-sulfated amino terminus have adaptive mutations in gp120, including loss of a functional N-glycan.J Virol. 2005 Apr;79(7):4357-68. doi: 10.1128/JVI.79.7.4357-4368.2005. J Virol. 2005. PMID: 15767436 Free PMC article.
-
Reversible and efficient activation of HIV-1 cell entry by a tyrosine-sulfated peptide dissects endocytic entry and inhibitor mechanisms.J Virol. 2014 Apr;88(8):4304-18. doi: 10.1128/JVI.03447-13. Epub 2014 Jan 29. J Virol. 2014. PMID: 24478426 Free PMC article.
-
Molecular Mechanism of HIV-1 Entry.Trends Microbiol. 2019 Oct;27(10):878-891. doi: 10.1016/j.tim.2019.06.002. Epub 2019 Jun 28. Trends Microbiol. 2019. PMID: 31262533 Free PMC article. Review.
-
CD4 activation of HIV fusion.Int J Cell Cloning. 1992 Nov;10(6):323-32. doi: 10.1002/stem.5530100603. Int J Cell Cloning. 1992. PMID: 1281202 Review.
Cited by
-
Adaptive mutations in a human immunodeficiency virus type 1 envelope protein with a truncated V3 loop restore function by improving interactions with CD4.J Virol. 2009 Nov;83(21):11005-15. doi: 10.1128/JVI.01238-09. Epub 2009 Aug 19. J Virol. 2009. PMID: 19692476 Free PMC article.
-
Mechanisms of receptor/coreceptor-mediated entry of enveloped viruses.Biophys J. 2009 Apr 8;96(7):2624-36. doi: 10.1016/j.bpj.2009.01.018. Biophys J. 2009. PMID: 19348746 Free PMC article.
-
Structures and mechanisms of viral membrane fusion proteins: multiple variations on a common theme.Crit Rev Biochem Mol Biol. 2008 May-Jun;43(3):189-219. doi: 10.1080/10409230802058320. Crit Rev Biochem Mol Biol. 2008. PMID: 18568847 Free PMC article. Review.
-
Inefficient entry of vicriviroc-resistant HIV-1 via the inhibitor-CCR5 complex at low cell surface CCR5 densities.Virology. 2009 May 10;387(2):296-302. doi: 10.1016/j.virol.2009.02.044. Epub 2009 Mar 20. Virology. 2009. PMID: 19303620 Free PMC article.
-
Kinetic mechanism for HIV-1 neutralization by antibody 2G12 entails reversible glycan binding that slows cell entry.Proc Natl Acad Sci U S A. 2012 May 15;109(20):7829-34. doi: 10.1073/pnas.1109728109. Epub 2012 Apr 30. Proc Natl Acad Sci U S A. 2012. PMID: 22547820 Free PMC article.
References
-
- Eckert DM, Kim PS. Mechanisms of viral membrane fusion and its inhibition. Annu Rev Biochem. 2001;70:777–810. - PubMed
-
- Wyatt R, Sodroski J. The HIV-1 envelope glycoproteins: fusogens, antigens, and immunogens. Science. 1998;280:1884–8. - PubMed
-
- Derdeyn CA, Decker JM, Sfakianos JN, Zhang Z, O’Brien WA, Ratner L, Shaw GM, Hunter E. Sensitivity of human immunodeficiency virus type 1 to fusion inhibitors targeted to the gp41 first heptad repeat involves distinct regions of gp41 and Is consistently modulated by gp120 interactions with the coreceptor. Journal of Virology. 2001;75:8605–14. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
