

Understanding the accuracy of statistical haplotype inference with sequence data of known phase
- PMID: 17922479
- PMCID: PMC2291540
- DOI: 10.1002/gepi.20185
Understanding the accuracy of statistical haplotype inference with sequence data of known phase
Abstract
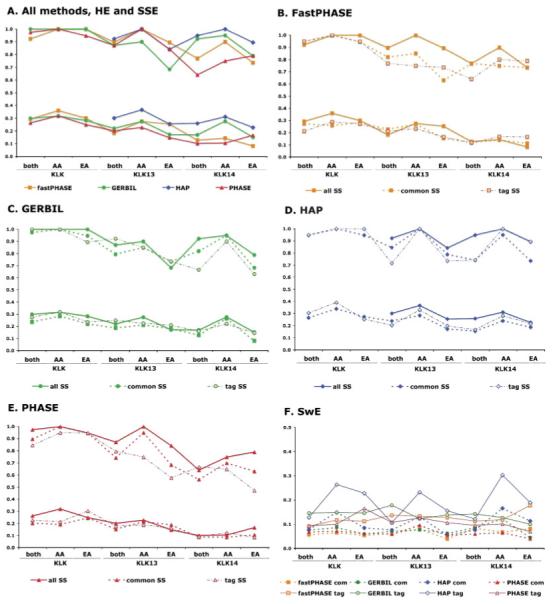
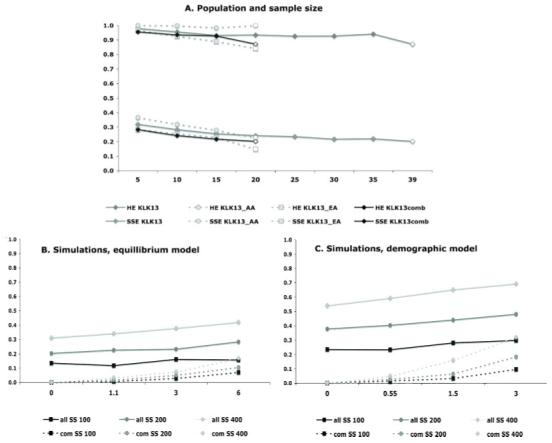
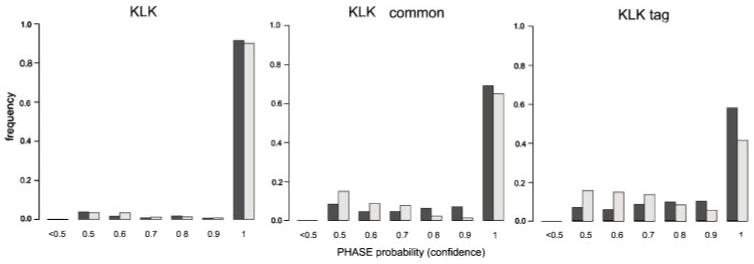
Statistical methods for haplotype inference from multi-site genotypes of unrelated individuals have important application in association studies and population genetics. Understanding the factors that affect the accuracy of this inference is important, but their assessment has been restricted by the limited availability of biological data with known phase. We created hybrid cell lines monosomic for human chromosome 19 and produced single-chromosome complete sequences of a 48 kb genomic region in 39 individuals of African American (AA) and European American (EA) origin. We employ these phase-known genotypes and coalescent simulations to assess the accuracy of statistical haplotype reconstruction by several algorithms. Accuracy of phase inference was considerably low in our biological data even for regions as short as 25-50 kb, suggesting that caution is needed when analyzing reconstructed haplotypes. Moreover, the reliability of estimated confidence in phase inference is not high enough to allow for a reliable incorporation of site-specific uncertainty information in subsequent analyses. We show that, in samples of certain mixed ancestry (AA and EA populations), the most accurate haplotypes are probably obtained when increasing sample size by considering the largest, pooled sample, despite the hypothetical problems associated with pooling across those heterogeneous samples. Strategies to improve confidence in reconstructed haplotypes, and realistic alternatives to the analysis of inferred haplotypes, are discussed.
Figures
References
-
- Barrett JC, Fry B, Maller J, Daly MJ, Barrett JC, Fry B, Maller J, Daly MJ. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics. 2005;21:263–265. - PubMed
-
- Chung RH, Gusfield D. Perfect phylogeny haplotyper: haplotype inferral using a tree model. Bioinformatics. 2003;19:780–781. - PubMed
-
- Clark AG. Inference of haplotypes from PCR-amplified samples of diploid populations. Mol Biol Evol. 1990;7:111–122. - PubMed
-
- Clark AG. The role of haplotypes in candidate gene studies. Genet Epidemiol. 2004;27:321–333. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
