

A patient with bilateral pheochromocytoma as part of a Von Hippel-Lindau (VHL) syndrome type 2C
- PMID: 17922902
- PMCID: PMC2169240
- DOI: 10.1186/1477-7819-5-112
A patient with bilateral pheochromocytoma as part of a Von Hippel-Lindau (VHL) syndrome type 2C
Abstract
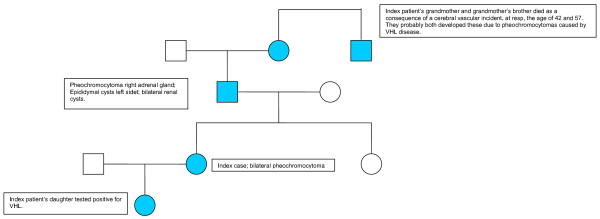
Background: Von Hippel-Lindau (VHL) disease is an autosomal dominant inherited disease. It is relatively recent that type 2C was identified as a separate group solely presenting with pheochromocytomas. As an illustration, an interesting case is presented of a pregnant woman with refractory hypertension. It proved to be the first manifestation of bilateral pheochromocytomas. The family history may indicate the diagnosis, but only identification of a germ line mutation in the DNA of a patient will confirm carriership.
Case presentation: A 27 year pregnant patient with intra uterine growth retardation presented with hypertension and pre-eclampsia. Magnetic resonance imaging revealed bilateral adrenal pheochromocytoma. She underwent laparoscopic adrenelectomy and a missense mutation (Gly93Ser) in exon 1 of the VHL gene on chromosome 3 (p25 - p26) was shown in the patient, her father and her daughter confirming the diagnosis of VHL.
Conclusion: In almost all VHL families molecular genetic analysis of DNA will demonstrate an inherited mutation. Because of the involvement in several organs, periodic clinical evaluation should take place in a well coordinated, multidisciplinary setting. VHL disease can be classified into several subtypes. VHL type 2C patients present with pheochromocytomas without evidence of haemangioblastomas in the central nervous system and/or retina and a low risk of renal cell carcinoma. Therefore, in such families, periodic clinical screening can be focussed on pheochromocytomas.
References
-
- Amar L, Bertherat J, Baudin E, Ajzenberg C, Bressac-de Paillerets B, Chabre O, Chamontin B, Delemer B, Giraud S, Murat A, Niccoli-Sire P, Richard S, Rohmer V, Sadoul JL, Strompf L, Schlumberger M, Bertagna X, Plouin PF, Jeunemaitre X, Gimenez-Roqueplo AP. Genetic testing in pheochromocytoma or functional paraganglioma. J Clin Oncol. 2005;23:8812–8818. doi: 10.1200/JCO.2005.03.1484. - DOI - PubMed
-
- Kebebew E, Duh QY. Benign and malignant pheochromocytoma: diagnosis, treatment, and follow-Up. Surg Oncol Clin N Am. 1998;7:765–789. - PubMed
-
- Neumann HP, Bausch B, McWhinney SR, Bender BU, Gimm O, Franke G, Schipper J, Klisch J, Altehoefer C, Zerres K, Januszewicz A, Eng C, Smith WM, Munk R, Manz T, Glaesker S, Apel TW, Treier M, Reineke M, Walz MK, Hoang-Vu C, Brauckhoff M, Klein-Franke A, Klose P, Schmidt H, Maier-Woelfle M, Peczkowska M, Szmigielski C, Eng C. Germ-line mutations in nonsyndromic pheochromocytoma. N Engl J Med. 2002;346:1459–1466. doi: 10.1056/NEJMoa020152. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
Research Materials
