

Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor {gamma} and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy
- PMID: 17932108
- PMCID: PMC2234580
- DOI: 10.1210/me.2007-0124
Human immunodeficiency virus (HIV)-1 viral protein R suppresses transcriptional activity of peroxisome proliferator-activated receptor {gamma} and inhibits adipocyte differentiation: implications for HIV-associated lipodystrophy
Abstract
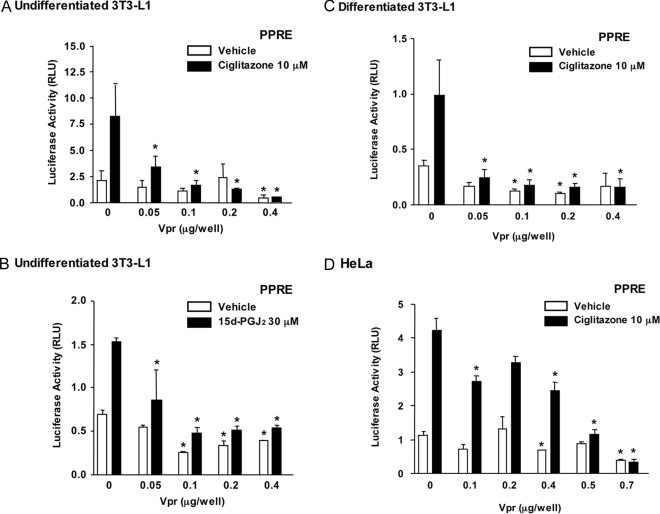
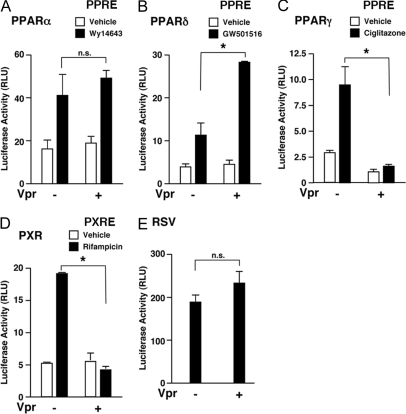
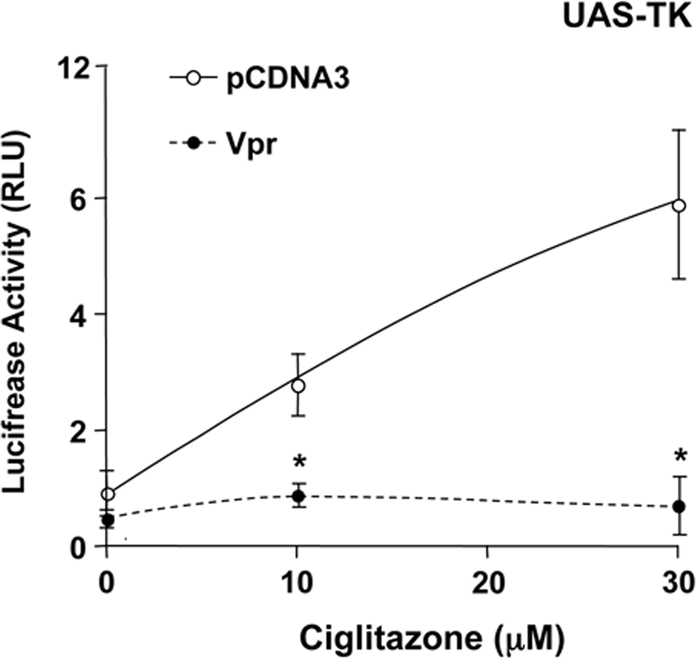
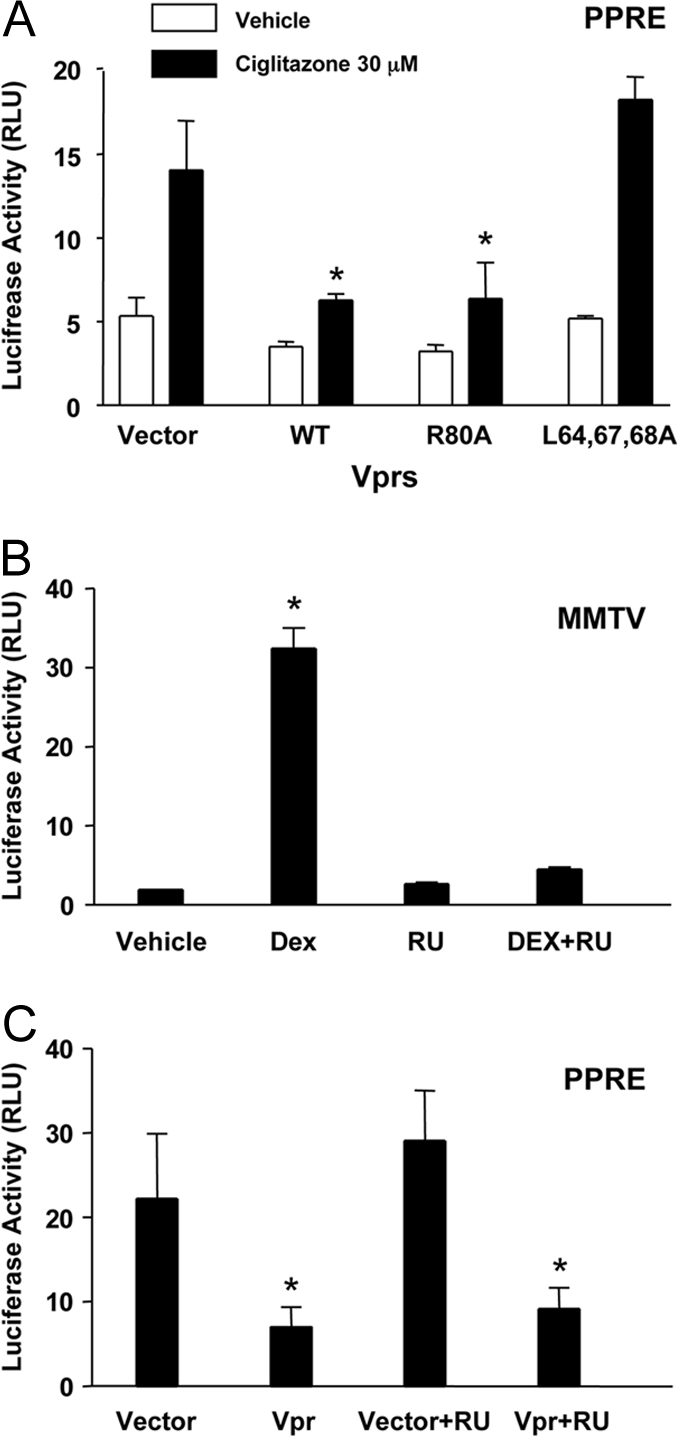
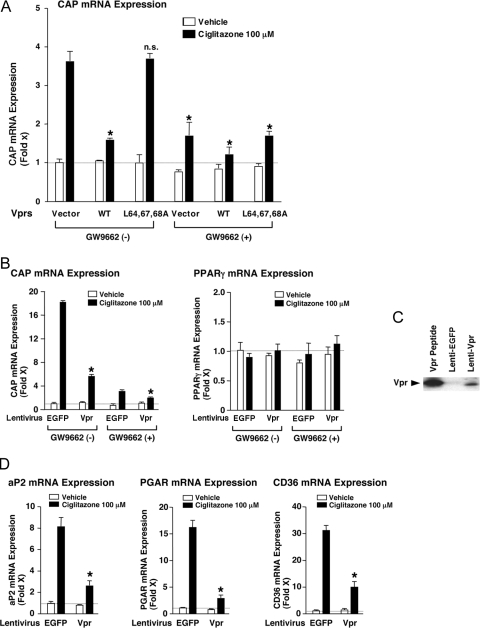
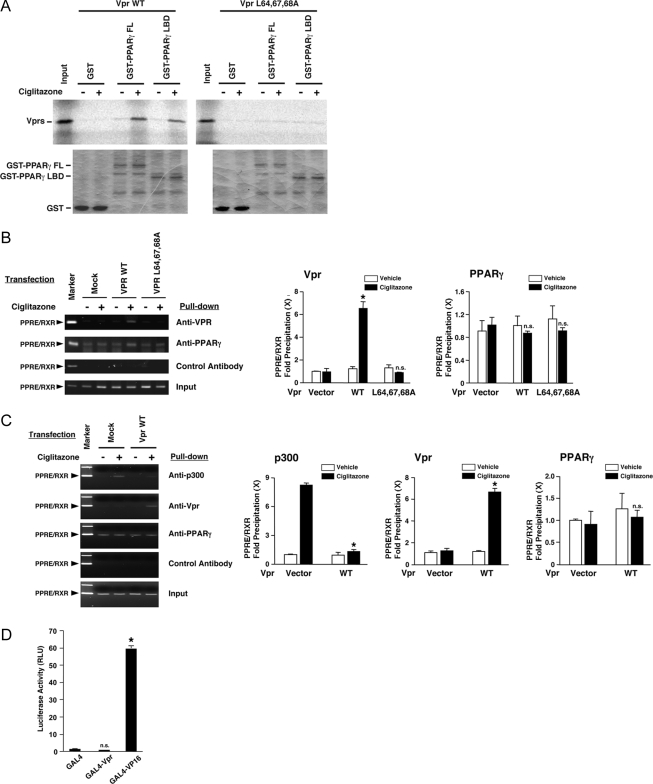
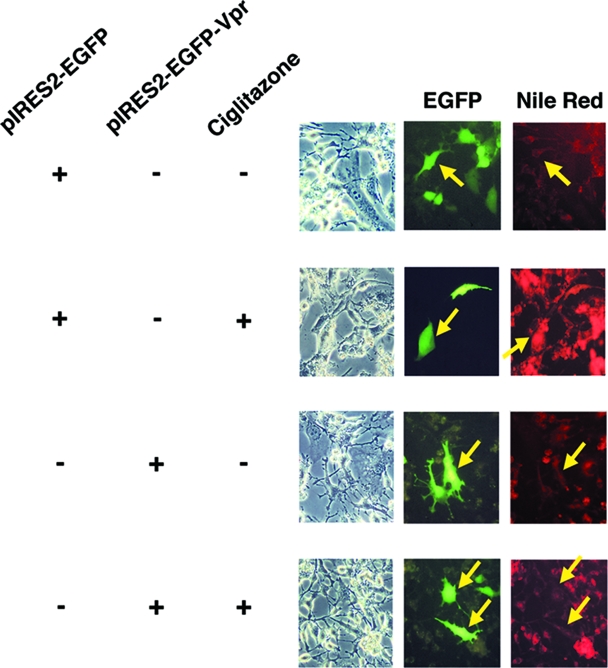
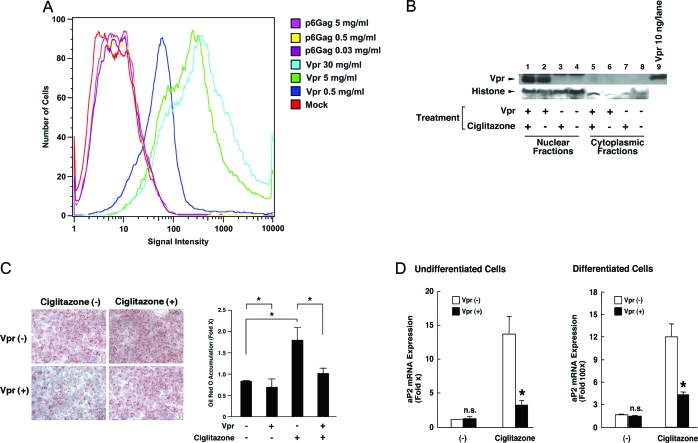
HIV-1-infected patients may develop lipodystrophy and insulin resistance. We investigated the effect of the HIV-1 accessory protein viral protein R (Vpr) on the activity of the peroxisome proliferator-activating receptor-gamma (PPARgamma), a key regulator of adipocyte differentiation and tissue insulin sensitivity. We studied expression of PPARgamma-responsive reporter genes in 3T3-L1 mouse adipocytes. We investigated Vpr interaction with the PPAR/retinoid X receptor (RXR)-binding site of the c-Cbl-associating protein (CAP) gene using the chromatin immunoprecipitation assay as well as the interaction of Vpr and PPARgamma using coimmunoprecipitation. Finally, we studied the ability of exogenous Vpr protein to enter cultured adipocytes and retard differentiation. We found that Vpr suppressed PPARgamma-induced transactivation in both undifferentiated and differentiated 3T3-L1 cells. Transcriptional suppression by Vpr required an intact LXXLL coactivator motif. Vpr suppressed mRNA expression of PPARgamma-responsive genes in undifferentiated 3T3-L1 cells and associated with the PPAR/RXR-binding site located in the promoter region of the CAP gene. Vpr interacted with the ligand-binding domain of PPARgamma in an agonist-dependent fashion in vitro. Vpr delivered either by an expression plasmid or as protein added to media suppressed PPARgamma agonist-induced adipocyte differentiation, assessed as lipid accumulation and mRNA expression of the adipocyte differentiation marker adipocyte P2 in 3T3-L1 cells. In conclusion, circulating Vpr or, alternatively, Vpr produced as a consequence of direct infection of adipocytes could suppress in vivo differentiation of preadipocytes by acting as a corepressor of PPARgamma-mediated gene transcription. Vpr may alter sensitivity to insulin and thereby contribute to the development of lipodystrophy and insulin resistance observed in HIV-1-infected patients.
Figures
References
-
- Carr A, Samaras K, Burton S, Law M, Freund J, Chisholm DJ, Cooper DA 1998 A syndrome of peripheral lipodystrophy, hyperlipidaemia and insulin resistance in patients receiving HIV protease inhibitors. AIDS 12:F51–F58 - PubMed
-
- Sweet DE 2005 Metabolic complications of antiretroviral therapy. Top HIV Med 13:70–74 - PubMed
-
- Lichtenstein KA 2005 Redefining lipodystrophy syndrome: risks and impact on clinical decision making. J Acquir Immune Defic Syndr 39:395–400 - PubMed
-
- Rudich A, Ben-Romano R, Etzion S, Bashan N 2005 Cellular mechanisms of insulin resistance, lipodystrophy and atherosclerosis induced by HIV protease inhibitors. Acta Physiol Scand 183:75–88 - PubMed
-
- Boufassa F, Dulioust A, Lascaux AS, Meyer L, Boue F, Delfraissy JF, Sobel A, Goujard C 2001 Lipodystrophy in 685 HIV-1-treated patients: influence of antiretroviral treatment and immunovirological response. HIV Clin Trials 2:339–345 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
