

Separating the effects of mutation and selection in producing DNA skew in bacterial chromosomes
- PMID: 17935620
- PMCID: PMC2099444
- DOI: 10.1186/1471-2164-8-369
Separating the effects of mutation and selection in producing DNA skew in bacterial chromosomes
Abstract
Background: Many bacterial chromosomes display nucleotide asymmetry, or skew, between the leading and lagging strands of replication. Mutational differences between these strands result in an overall pattern of skew that is centered about the origin of replication. Such a pattern could also arise from selection coupled with a bias for genes coded on the leading strand. The relative contributions of selection and mutation in producing compositional skew are largely unknown.
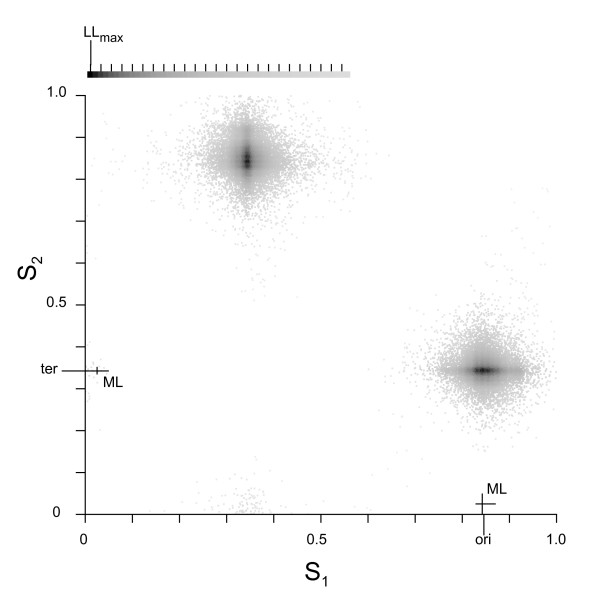
Results: We describe a model to quantify the contribution of mutational differences between the leading and lagging strands in producing replication-induced skew. When the origin and terminus of replication are known, the model can be used to estimate the relative accumulation of G over C and of A over T on the leading strand due to replication effects in a chromosome with bidirectional replication arms. The model may also be implemented in a maximum likelihood framework to estimate the locations of origin and terminus. We find that our estimations for the origin and terminus agree very well with the location of genes that are thought to be associated with the replication origin. This indicates that our model provides an accurate, objective method of determining the replication arms and also provides support for the hypothesis that these genes represent an ancestral cluster of origin-associated genes.
Conclusion: The model has several advantages over other methods of analyzing genome skew. First, it quantifies the role of mutation in generating skew so that its effect on composition, for example codon bias, can be assessed. Second, it provides an objective method for locating origin and terminus, one that is based on chromosome-wide accumulation of leading vs lagging strand nucleotide differences. Finally, the model has the potential to be utilized in a maximum likelihood framework in order to analyze the effect of chromosome rearrangements on nucleotide composition.
Figures
Similar articles
-
GC skew in protein-coding genes between the leading and lagging strands in bacterial genomes: new substitution models incorporating strand bias.J Theor Biol. 2008 Aug 7;253(3):508-13. doi: 10.1016/j.jtbi.2008.04.004. Epub 2008 Apr 11. J Theor Biol. 2008. PMID: 18486155
-
Quantitative analysis of replication-related mutation and selection pressures in bacterial chromosomes and plasmids using generalised GC skew index.BMC Genomics. 2009 Dec 30;10:640. doi: 10.1186/1471-2164-10-640. BMC Genomics. 2009. PMID: 20042086 Free PMC article.
-
Methods for comparing sources of strand compositional asymmetry in microbial chromosomes.DNA Res. 2003 Jun 30;10(3):85-95. doi: 10.1093/dnares/10.3.85. DNA Res. 2003. PMID: 12886951
-
Polarization of the Escherichia coli chromosome. A view from the terminus.Biochimie. 2001 Feb;83(2):161-70. doi: 10.1016/s0300-9084(00)01202-5. Biochimie. 2001. PMID: 11278065 Review.
-
Is there a role for replication fork asymmetry in the distribution of genes in bacterial genomes?Trends Microbiol. 2002 Sep;10(9):393-5. doi: 10.1016/s0966-842x(02)02420-4. Trends Microbiol. 2002. PMID: 12217498 Review.
Cited by
-
Atypical at skew in Firmicute genomes results from selection and not from mutation.PLoS Genet. 2011 Sep;7(9):e1002283. doi: 10.1371/journal.pgen.1002283. Epub 2011 Sep 15. PLoS Genet. 2011. PMID: 21935355 Free PMC article.
-
Mitochondrial Genome Variations and Possible Adaptive Implications in Some Tephritid Flies (Diptera, Tephritidae).Int J Mol Sci. 2025 Jun 10;26(12):5560. doi: 10.3390/ijms26125560. Int J Mol Sci. 2025. PMID: 40565023 Free PMC article.
-
Substitution rate heterogeneity across hexanucleotide contexts in noncoding chloroplast DNA.G3 (Bethesda). 2022 Jul 29;12(8):jkac150. doi: 10.1093/g3journal/jkac150. G3 (Bethesda). 2022. PMID: 35699494 Free PMC article.
-
Codon Adaptation of Plastid Genes.PLoS One. 2016 May 19;11(5):e0154306. doi: 10.1371/journal.pone.0154306. eCollection 2016. PLoS One. 2016. PMID: 27196606 Free PMC article.
-
Measures of compositional strand bias related to replication machinery and its applications.Curr Genomics. 2012 Mar;13(1):4-15. doi: 10.2174/138920212799034749. Curr Genomics. 2012. PMID: 22942671 Free PMC article.
References
-
- Blattner FR, Plunkett G, 3rd, Bloch CA, Perna NT, Burland V, Riley M, Collado-Vides J, Glasner JD, Rode CK, Mayhew GF, Gregor J, Davis NW, Kirkpatrick HA, Goeden MA, Rose DJ, Mau B, Shao Y. The Complete Genome Sequence of Escherichia coli K-12. Science. 1997;277:1453–1462. doi: 10.1126/science.277.5331.1453. - DOI - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
