

Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle
- PMID: 17937813
- PMCID: PMC2140053
- DOI: 10.1186/1471-2350-8-64
Metabolic phenotype of methylmalonic acidemia in mice and humans: the role of skeletal muscle
Abstract
Background: Mutations in methylmalonyl-CoA mutase cause methylmalonic acidemia, a common organic aciduria. Current treatment regimens rely on dietary management and, in severely affected patients, liver or combined liver-kidney transplantation. For undetermined reasons, transplantation does not correct the biochemical phenotype.
Methods: To study the metabolic disturbances seen in this disorder, we have created a murine model with a null allele at the methylmalonyl-CoA mutase locus and correlated the results observed in the knock-out mice to patient data. To gain insight into the origin and magnitude of methylmalonic acid (MMA) production in humans with methylmalonyl-CoA mutase deficiency, we evaluated two methylmalonic acidemia patients who had received different variants of combined liver-kidney transplants, one with a complete liver replacement-kidney transplant and the other with an auxiliary liver graft-kidney transplant, and compared their metabolite production to four untransplanted patients with intact renal function.
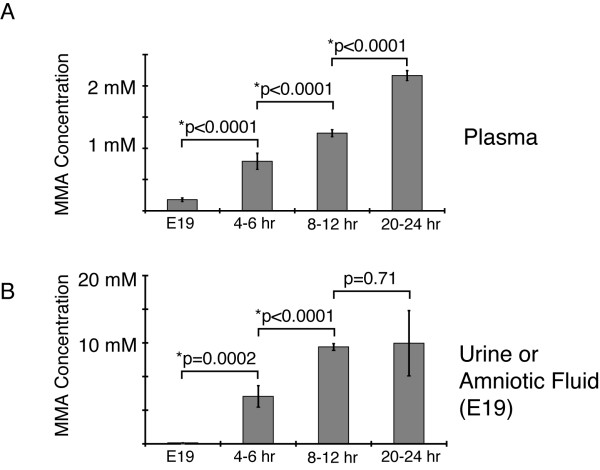
Results: Enzymatic, Western and Northern analyses demonstrated that the targeted allele was null and correctable by lentiviral complementation. Metabolite studies defined the magnitude and tempo of plasma MMA concentrations in the mice. Before a fatal metabolic crisis developed in the first 24-48 hours, the methylmalonic acid content per gram wet-weight was massively elevated in the skeletal muscle as well as the kidneys, liver and brain. Near the end of life, extreme elevations in tissue MMA were present primarily in the liver. The transplant patients studied when well and on dietary therapy, displayed massive elevations of MMA in the plasma and urine, comparable to the levels seen in the untransplanted patients with similar enzymatic phenotypes and dietary regimens.
Conclusion: The combined observations from the murine metabolite studies and patient investigations indicate that during homeostasis, a large portion of circulating MMA has an extra-heptorenal origin and likely derives from the skeletal muscle. Our studies suggest that modulating skeletal muscle metabolism may represent a strategy to increase metabolic capacity in methylmalonic acidemia as well as other organic acidurias. This mouse model will be useful for further investigations exploring disease mechanisms and therapeutic interventions in methylmalonic acidemia, a devastating disorder of intermediary metabolism.
Figures




References
-
- Fenton WA, Gravel RA, Rosenblatt DS. In: Disorders of Propionate and Methylmalonate Metabolism in The Metabolic and Molecular Bases for Inhertited Disease. 8. Scriver CR, Beaudet AL, Sly WS, Valle D, editor. McGraw-Hill, Inc., New York; 2001. pp. 2165–2192.
-
- Fenton WA, Rosenblatt DS. In: Inherited Disorders of Folate and Cobalamin Transport and Metabolism in The Metabolic and Molecular Bases of Inherited Disease. 8. Scriver CR BA, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B, editor. McGraw-Hill; 2001. pp. 3897–3933.
-
- Matsui SM, Mahoney MJ, Rosenberg LE. The natural history of the inherited methylmalonic acidemias. N Engl J Med. 1983;308:857–861. - PubMed
-
- van der Meer SB, Poggi F, Spada M, Bonnefont JP, Ogier H, Hubert P, Depondt E, Rapoport D, Rabier D, Charpentier C, et al. Clinical outcome of long-term management of patients with vitamin B12-unresponsive methylmalonic acidemia. J Pediatr. 1994;125:903–908. doi: 10.1016/S0022-3476(05)82005-0. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
