

Improved human disease candidate gene prioritization using mouse phenotype
- PMID: 17939863
- PMCID: PMC2194797
- DOI: 10.1186/1471-2105-8-392
Improved human disease candidate gene prioritization using mouse phenotype
Abstract
Background: The majority of common diseases are multi-factorial and modified by genetically and mechanistically complex polygenic interactions and environmental factors. High-throughput genome-wide studies like linkage analysis and gene expression profiling, tend to be most useful for classification and characterization but do not provide sufficient information to identify or prioritize specific disease causal genes.
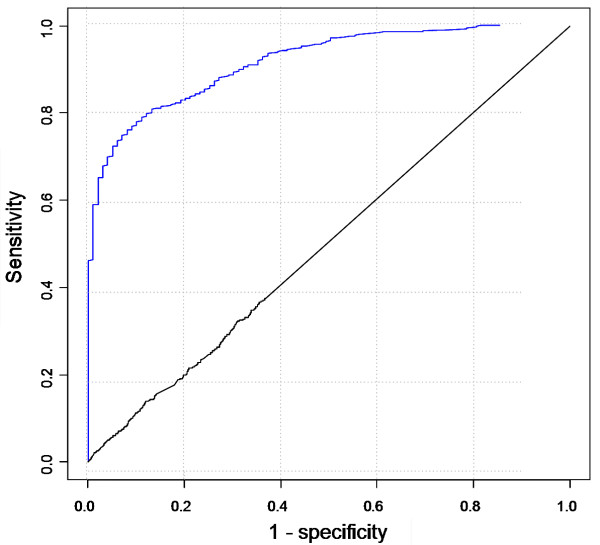
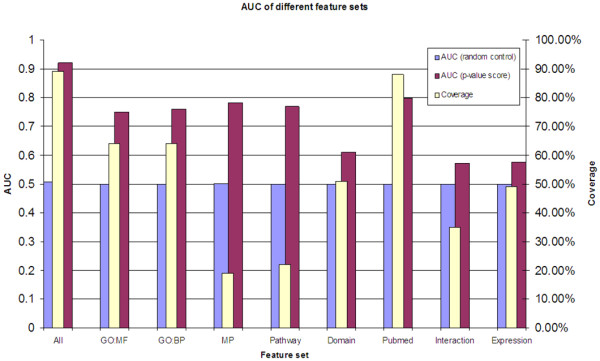
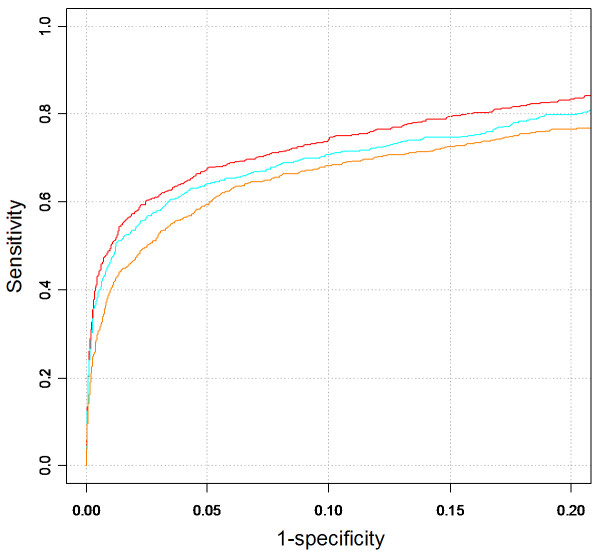
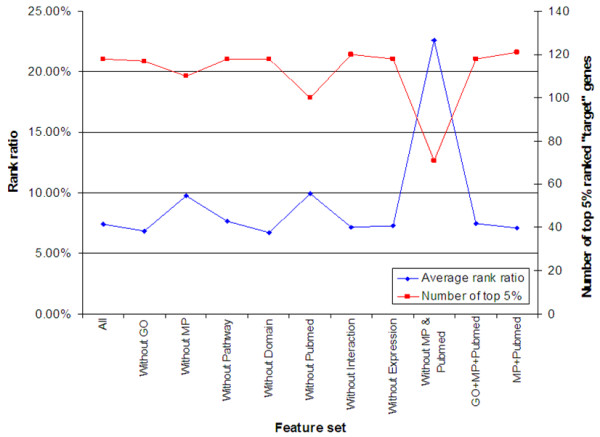
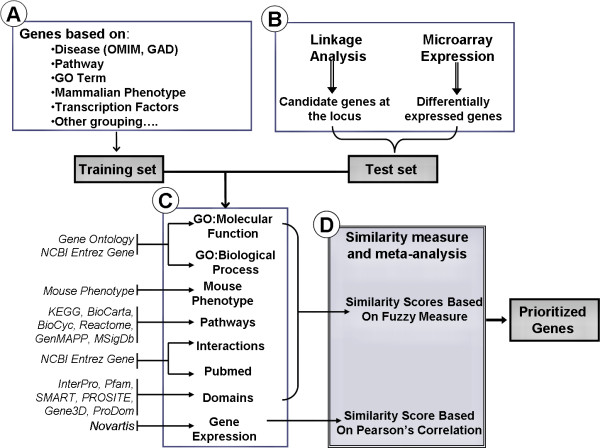
Results: Extending on an earlier hypothesis that the majority of genes that impact or cause disease share membership in any of several functional relationships we, for the first time, show the utility of mouse phenotype data in human disease gene prioritization. We study the effect of different data integration methods, and based on the validation studies, we show that our approach, ToppGene http://toppgene.cchmc.org, outperforms two of the existing candidate gene prioritization methods, SUSPECTS and ENDEAVOUR.
Conclusion: The incorporation of phenotype information for mouse orthologs of human genes greatly improves the human disease candidate gene analysis and prioritization.
Figures
References
-
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, Mesirov JP. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. doi: 10.1073/pnas.0506580102. - DOI - PMC - PubMed
-
- Freudenberg J, Propping P. A similarity-based method for genome-wide prediction of disease-relevant human genes. Bioinformatics. 2002;18 Suppl 2:S110–5. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
