

Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution
- PMID: 17939991
- PMCID: PMC2435059
- DOI: 10.1074/mcp.M700354-MCP200
Quantitative, multiplexed assays for low abundance proteins in plasma by targeted mass spectrometry and stable isotope dilution
Abstract
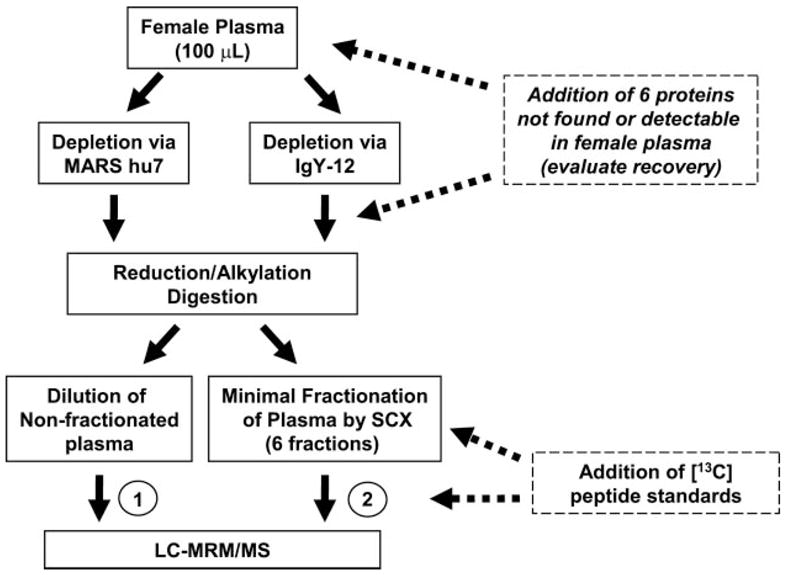
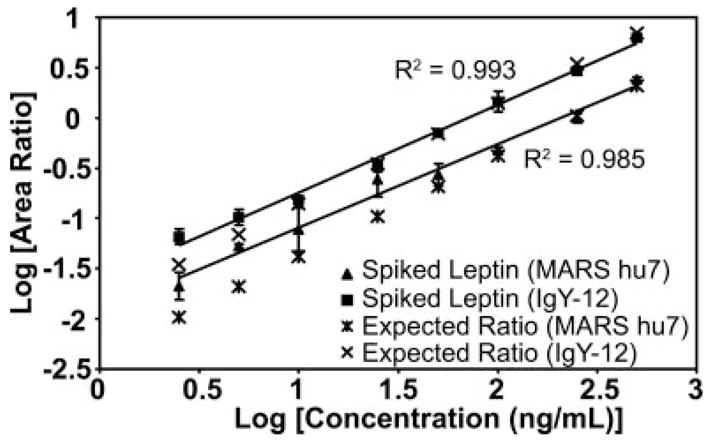
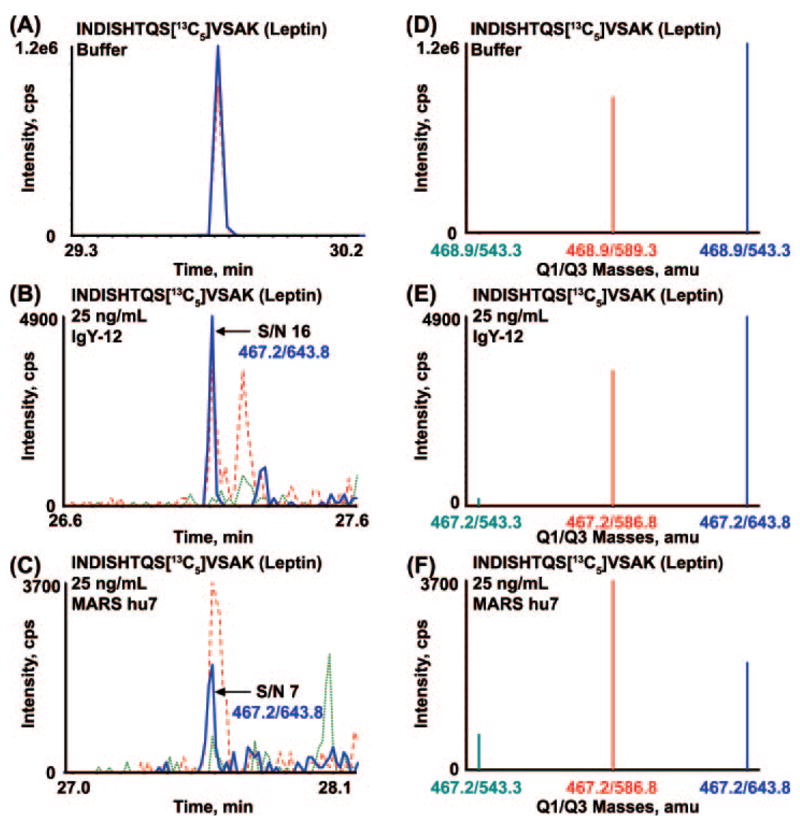
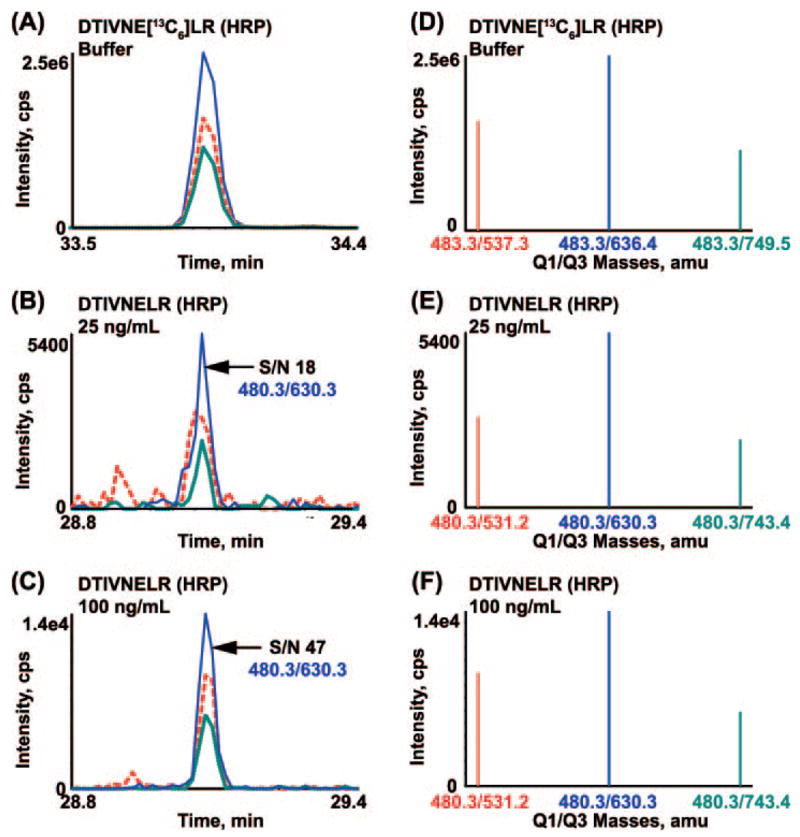
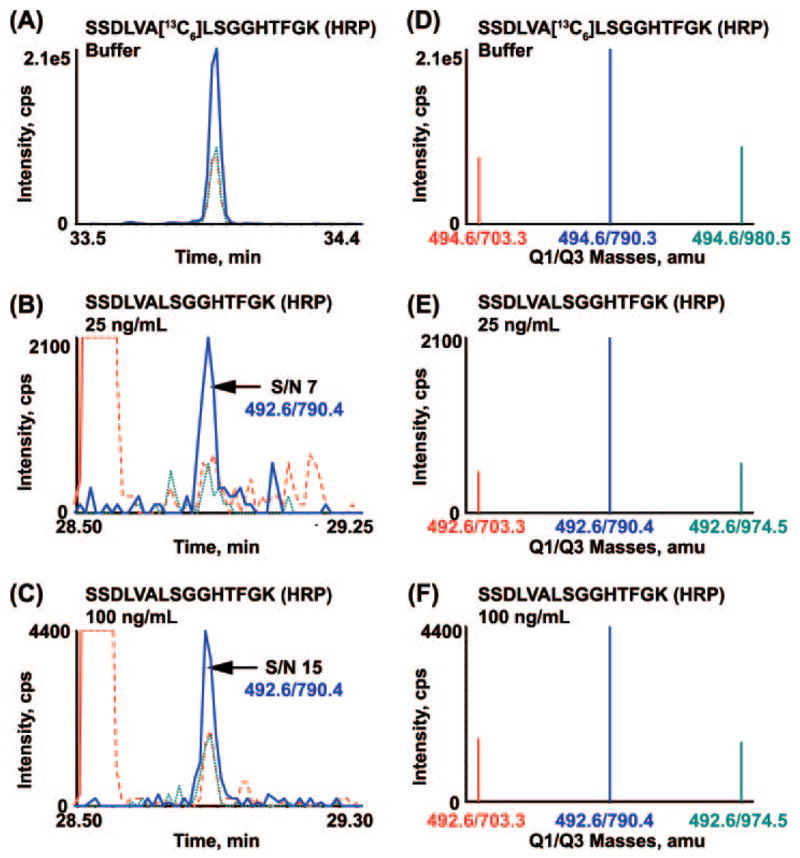
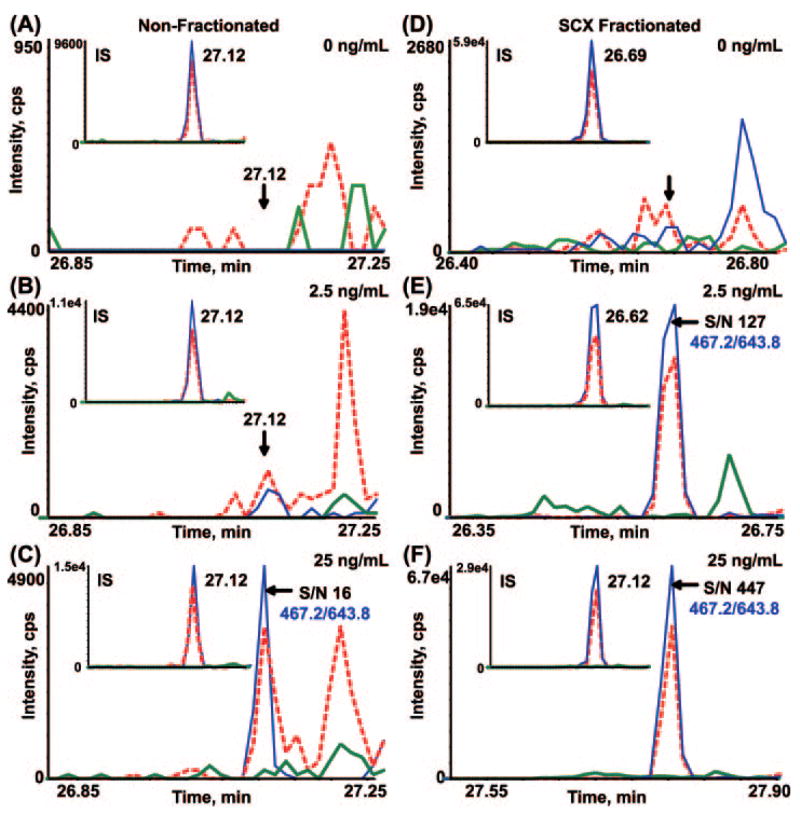
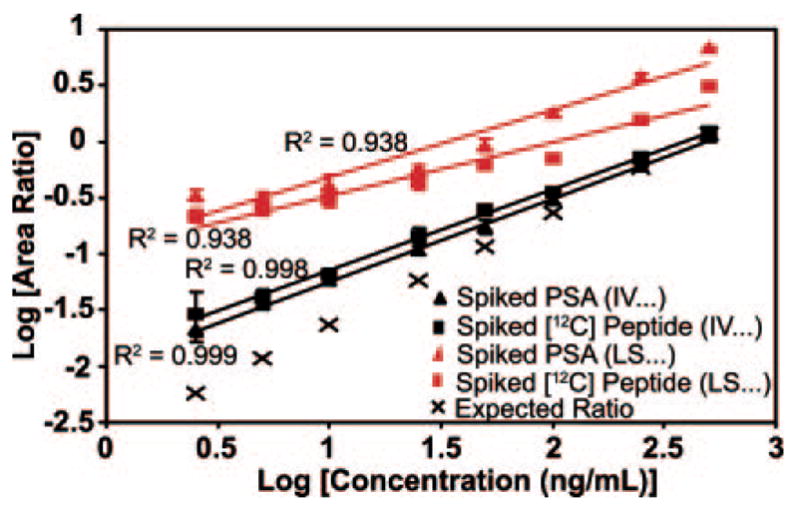
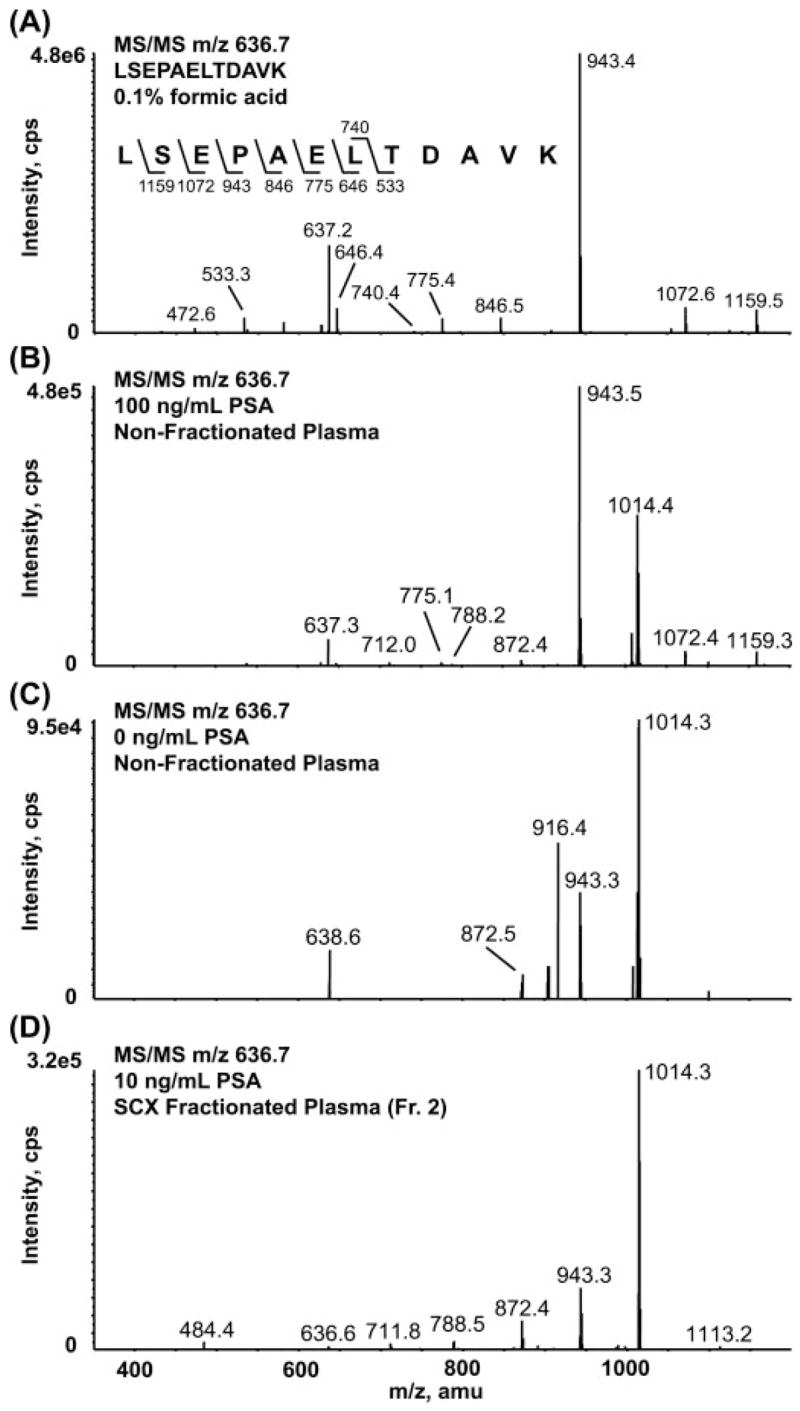
Biomarker discovery produces lists of candidate markers whose presence and level must be subsequently verified in serum or plasma. Verification represents a paradigm shift from unbiased discovery approaches to targeted, hypothesis-driven methods and relies upon specific, quantitative assays optimized for the selective detection of target proteins. Many protein biomarkers of clinical currency are present at or below the nanogram/milliliter range in plasma and have been inaccessible to date by MS-based methods. Using multiple reaction monitoring coupled with stable isotope dilution mass spectrometry, we describe here the development of quantitative, multiplexed assays for six proteins in plasma that achieve limits of quantitation in the 1-10 ng/ml range with percent coefficients of variation from 3 to 15% without immunoaffinity enrichment of either proteins or peptides. Sample processing methods with sufficient throughput, recovery, and reproducibility to enable robust detection and quantitation of candidate biomarker proteins were developed and optimized by addition of exogenous proteins to immunoaffinity depleted plasma from a healthy donor. Quantitative multiple reaction monitoring assays were designed and optimized for signature peptides derived from the test proteins. Based upon calibration curves using known concentrations of spiked protein in plasma, we determined that each target protein had at least one signature peptide with a limit of quantitation in the 1-10 ng/ml range and linearity typically over 2 orders of magnitude in the measurement range of interest. Limits of detection were frequently in the high picogram/milliliter range. These levels of assay performance represent up to a 1000-fold improvement compared with direct analysis of proteins in plasma by MS and were achieved by simple, robust sample processing involving abundant protein depletion and minimal fractionation by strong cation exchange chromatography at the peptide level prior to LC-multiple reaction monitoring/MS. The methods presented here provide a solid basis for developing quantitative MS-based assays of low level proteins in blood.
Figures
References
-
- Aebersold R, Mann M. Mass spectrometry-based proteomics. Nature. 2003;422:198–207. - PubMed
-
- Adkins JN, Varnum SM, Auberry KJ, Moore RJ, Angell NH, Smith RD, Springer DL, Pounds JG. Toward a human blood serum proteome: analysis by multidimensional separation coupled with mass spectrometry. Mol Cell Proteomics. 2002;1:947–955. - PubMed
-
- Rifai N, Gillette MA, Carr SA. Protein biomarker discovery and validation: the long and uncertain path to clinical utility. Nat Biotechnol. 2006;16:1929–1935. - PubMed
-
- Wang W, Zhou H, Lin H, Roy S, Shaler TA, Hill LR, Norton S, Kumar P, Anderle M, Becker CH. Quantification of proteins and metabolites by mass spectrometry without isotopic labeling or spiked standards. Anal Chem. 2003;75:4818–4826. - PubMed
-
- MacCoss MJ, Wu CC, Liu H, Sadygov R, Yates JR., III A correlation algorithm for the automated quantitative analysis of shotgun proteomics data. Anal Chem. 2003;75:6912–6921. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
