

Module-based outcome prediction using breast cancer compendia
- PMID: 17940611
- PMCID: PMC2002511
- DOI: 10.1371/journal.pone.0001047
Module-based outcome prediction using breast cancer compendia
Abstract
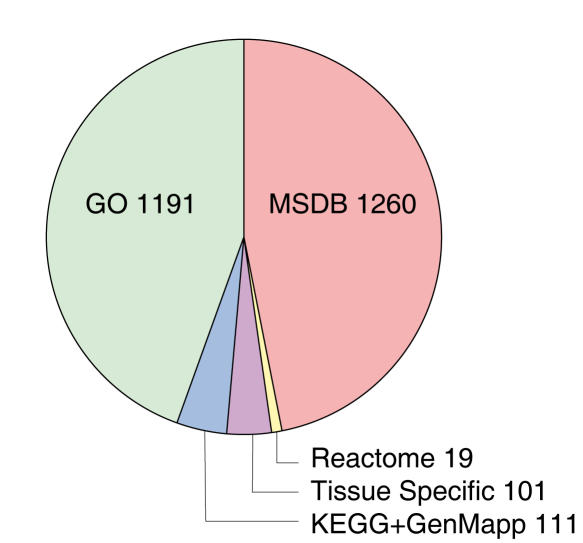
Background: The availability of large collections of microarray datasets (compendia), or knowledge about grouping of genes into pathways (gene sets), is typically not exploited when training predictors of disease outcome. These can be useful since a compendium increases the number of samples, while gene sets reduce the size of the feature space. This should be favorable from a machine learning perspective and result in more robust predictors.
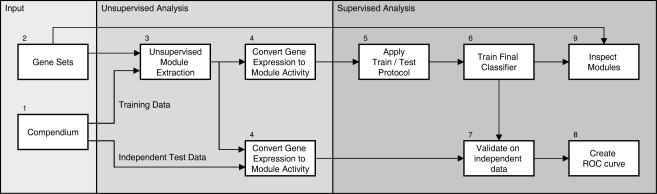
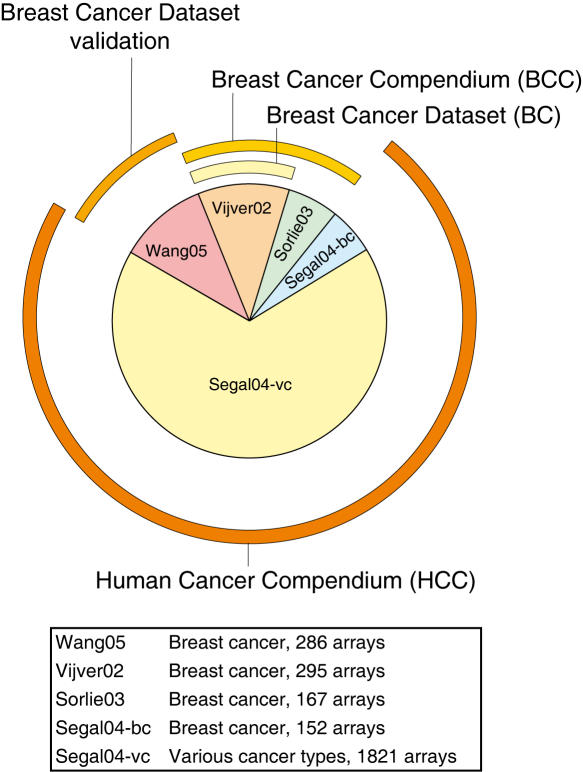
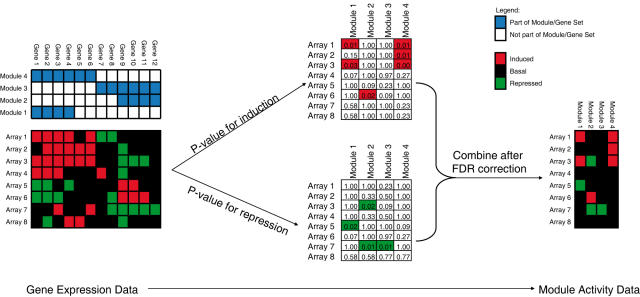
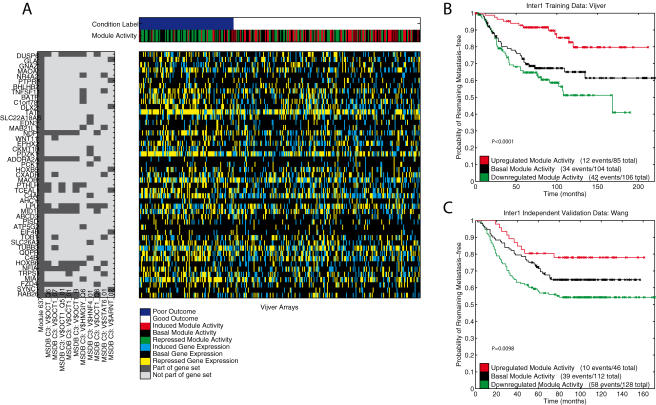
Methodology: We extracted modules of regulated genes from gene sets, and compendia. Through supervised analysis, we constructed predictors which employ modules predictive of breast cancer outcome. To validate these predictors we applied them to independent data, from the same institution (intra-dataset), and other institutions (inter-dataset).
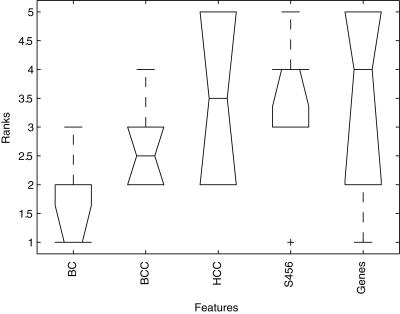
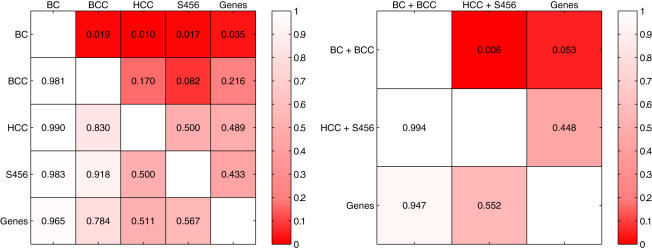
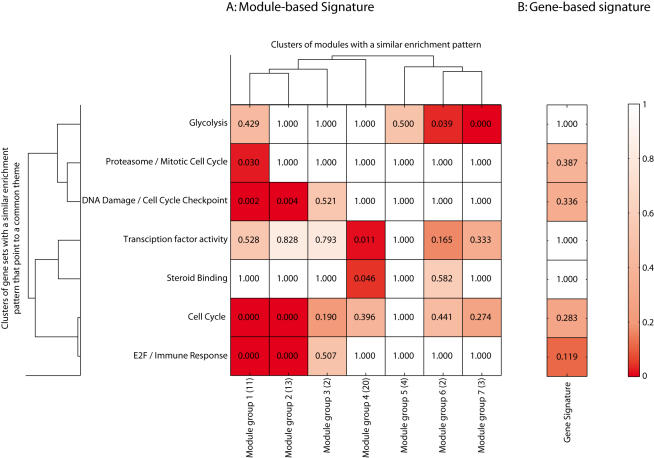
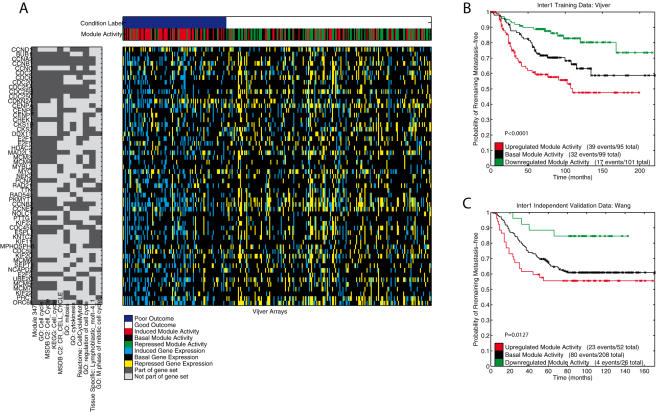
Conclusions: We show that modules derived from single breast cancer datasets achieve better performance on the validation data compared to gene-based predictors. We also show that there is a trend in compendium specificity and predictive performance: modules derived from a single breast cancer dataset, and a breast cancer specific compendium perform better compared to those derived from a human cancer compendium. Additionally, the module-based predictor provides a much richer insight into the underlying biology. Frequently selected gene sets are associated with processes such as cell cycle, E2F regulation, DNA damage response, proteasome and glycolysis. We analyzed two modules related to cell cycle, and the OCT1 transcription factor, respectively. On an individual basis, these modules provide a significant separation in survival subgroups on the training and independent validation data.
Conflict of interest statement
Figures
Similar articles
-
Prediction potential of candidate biomarker sets identified and validated on gene expression data from multiple datasets.BMC Bioinformatics. 2007 Oct 26;8:415. doi: 10.1186/1471-2105-8-415. BMC Bioinformatics. 2007. PMID: 17963508 Free PMC article.
-
Mixture classification model based on clinical markers for breast cancer prognosis.Artif Intell Med. 2010 Feb-Mar;48(2-3):129-37. doi: 10.1016/j.artmed.2009.07.008. Epub 2009 Dec 14. Artif Intell Med. 2010. PMID: 20005686
-
Integrating biological knowledge with gene expression profiles for survival prediction of cancer.J Comput Biol. 2009 Feb;16(2):265-78. doi: 10.1089/cmb.2008.12TT. J Comput Biol. 2009. PMID: 19183004 Free PMC article.
-
Molecular profiling of breast cancer: transcriptomic studies and beyond.Cell Mol Life Sci. 2007 Dec;64(24):3185-200. doi: 10.1007/s00018-007-7387-1. Cell Mol Life Sci. 2007. PMID: 17957338 Free PMC article. Review.
-
Using genome-wide expression compendia to study microorganisms.Comput Struct Biotechnol J. 2022 Aug 10;20:4315-4324. doi: 10.1016/j.csbj.2022.08.012. eCollection 2022. Comput Struct Biotechnol J. 2022. PMID: 36016717 Free PMC article. Review.
Cited by
-
Prediction of breast cancer prognosis using gene set statistics provides signature stability and biological context.BMC Bioinformatics. 2010 May 25;11:277. doi: 10.1186/1471-2105-11-277. BMC Bioinformatics. 2010. PMID: 20500821 Free PMC article.
-
Improved prognostic classification of breast cancer defined by antagonistic activation patterns of immune response pathway modules.BMC Cancer. 2010 Nov 4;10:604. doi: 10.1186/1471-2407-10-604. BMC Cancer. 2010. PMID: 21050467 Free PMC article.
-
Identifying cancer prognostic modules by module network analysis.BMC Bioinformatics. 2019 Feb 18;20(1):85. doi: 10.1186/s12859-019-2674-z. BMC Bioinformatics. 2019. PMID: 30777030 Free PMC article.
-
A computational model to predict bone metastasis in breast cancer by integrating the dysregulated pathways.BMC Cancer. 2014 Aug 27;14:618. doi: 10.1186/1471-2407-14-618. BMC Cancer. 2014. PMID: 25163697 Free PMC article.
-
Ensemble classifier based on context specific miRNA regulation modules: a new method for cancer outcome prediction.BMC Bioinformatics. 2013;14 Suppl 12(Suppl 12):S6. doi: 10.1186/1471-2105-14-S12-S6. Epub 2013 Sep 24. BMC Bioinformatics. 2013. PMID: 24268063 Free PMC article.
References
-
- Van 't Veer L, Dai H, van de Vijver M, He Y, Hart A, et al. Gene expression profiling predicts clinical outcome of breast cancer. Nature. 2002;415:530–6. - PubMed
-
- Wang Y, Klein J, Zhang Y, Sieuwerts A, Look M, et al. Gene-expression profiles to predict distant metastasis of lymph-node-negative primary breast cancer. The Lancet. 2005;365:671–9. - PubMed
-
- Van de Vijver M, He Y, van 't Veer L, Dai H, Hart A, et al. A gene-expression signature as a predictor of survival in breast cancer. N. Engl. J. Med. 2002;25:1999–2009. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
