

Structural analysis of kinetic folding intermediates for a TIM barrel protein, indole-3-glycerol phosphate synthase, by hydrogen exchange mass spectrometry and Gō model simulation
- PMID: 17942114
- PMCID: PMC2735044
- DOI: 10.1016/j.jmb.2007.09.024
Structural analysis of kinetic folding intermediates for a TIM barrel protein, indole-3-glycerol phosphate synthase, by hydrogen exchange mass spectrometry and Gō model simulation
Abstract
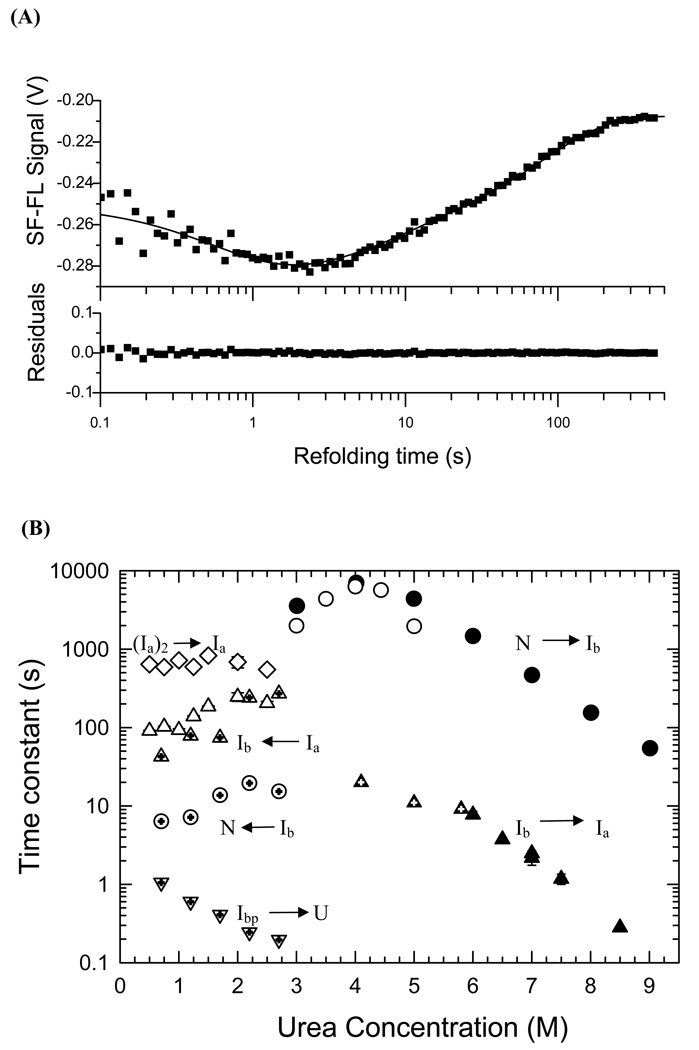
The structures of partially folded states appearing during the folding of a (betaalpha)(8) TIM barrel protein, the indole-3-glycerol phosphate synthase from Sulfolobus solfataricus (sIGPS), was assessed by hydrogen exchange mass spectrometry (HX-MS) and Gō model simulations. HX-MS analysis of the peptic peptides derived from the pulse-labeled product of the sub-millisecond folding reaction from the urea-denatured state revealed strong protection in the (betaalpha)(4) region, modest protection in the neighboring (betaalpha)(1-3) and (betaalpha)(5)beta(6) segments and no significant protection in the remaining N and C-terminal segments. These results demonstrate that this species is not a collapsed form of the unfolded state under native-favoring conditions nor is it the native state formed via fast-track folding. However, the striking contrast of these results with the strong protection observed in the (betaalpha)(2-5)beta(6) region after 5 s of folding demonstrates that these species represent kinetically distinct folding intermediates that are not identical as previously thought. A re-examination of the kinetic folding mechanism by chevron analysis of fluorescence data confirmed distinct roles for these two species: the burst-phase intermediate is predicted to be a misfolded, off-pathway intermediate, while the subsequent 5 s intermediate corresponds to an on-pathway equilibrium intermediate. Comparison with the predictions using a C(alpha) Gō model simulation of the kinetic folding reaction for sIGPS shows good agreement with the core of the structure offering protection against exchange in the on-pathway intermediate(s). Because the native-centric Gō model simulations do not explicitly include sequence-specific information, the simulation results support the hypothesis that the topology of TIM barrel proteins is a primary determinant of the folding free energy surface for the productive folding reaction. The early misfolding reaction must involve aspects of non-native structure not detected by the Gō model simulation.
Figures











Similar articles
-
Mapping the structure of folding cores in TIM barrel proteins by hydrogen exchange mass spectrometry: the roles of motif and sequence for the indole-3-glycerol phosphate synthase from Sulfolobus solfataricus.J Mol Biol. 2007 Apr 27;368(2):582-94. doi: 10.1016/j.jmb.2007.02.027. Epub 2007 Feb 20. J Mol Biol. 2007. PMID: 17359995 Free PMC article.
-
Folding mechanism of indole-3-glycerol phosphate synthase from Sulfolobus solfataricus: a test of the conservation of folding mechanisms hypothesis in (beta(alpha))(8) barrels.J Mol Biol. 2002 Jul 26;320(5):1119-33. doi: 10.1016/s0022-2836(02)00557-0. J Mol Biol. 2002. PMID: 12126630
-
Topology and sequence in the folding of a TIM barrel protein: global analysis highlights partitioning between transient off-pathway and stable on-pathway folding intermediates in the complex folding mechanism of a (betaalpha)8 barrel of unknown function from B. subtilis.J Mol Biol. 2007 Sep 7;372(1):236-53. doi: 10.1016/j.jmb.2007.06.018. Epub 2007 Jun 14. J Mol Biol. 2007. PMID: 17619021
-
Hydrogen exchange methods to study protein folding.Methods. 2004 Sep;34(1):51-64. doi: 10.1016/j.ymeth.2004.03.005. Methods. 2004. PMID: 15283915 Review.
-
Folding of apomyoglobin: Analysis of transient intermediate structure during refolding using quick hydrogen deuterium exchange and NMR.Proc Jpn Acad Ser B Phys Biol Sci. 2017;93(1):10-27. doi: 10.2183/pjab.93.002. Proc Jpn Acad Ser B Phys Biol Sci. 2017. PMID: 28077807 Free PMC article. Review.
Cited by
-
Clusters of branched aliphatic side chains serve as cores of stability in the native state of the HisF TIM barrel protein.J Mol Biol. 2013 Mar 25;425(6):1065-81. doi: 10.1016/j.jmb.2013.01.002. Epub 2013 Jan 16. J Mol Biol. 2013. PMID: 23333740 Free PMC article.
-
A conserved folding nucleus sculpts the free energy landscape of bacterial and archaeal orthologs from a divergent TIM barrel family.Proc Natl Acad Sci U S A. 2021 Apr 27;118(17):e2019571118. doi: 10.1073/pnas.2019571118. Proc Natl Acad Sci U S A. 2021. PMID: 33875592 Free PMC article.
-
Insights from coarse-grained Gō models for protein folding and dynamics.Int J Mol Sci. 2009 Mar;10(3):889-905. doi: 10.3390/ijms10030889. Epub 2009 Mar 2. Int J Mol Sci. 2009. PMID: 19399227 Free PMC article. Review.
-
Validation of DBFOLD: An efficient algorithm for computing folding pathways of complex proteins.PLoS Comput Biol. 2020 Nov 16;16(11):e1008323. doi: 10.1371/journal.pcbi.1008323. eCollection 2020 Nov. PLoS Comput Biol. 2020. PMID: 33196646 Free PMC article.
-
Protein dynamics governed by interfaces of high polarity and low packing density.PLoS One. 2012;7(10):e48212. doi: 10.1371/journal.pone.0048212. Epub 2012 Oct 26. PLoS One. 2012. PMID: 23110216 Free PMC article.
References
-
- Jackson SE. How do small single-domain proteins fold? Fold Des. 1998;3:R81–R91. - PubMed
-
- Ballew RM, Sabelko J, Gruebele M. Observation of distinct nanosecond and microsecond protein folding events. Nat Struct Biol. 1996;3:923–926. - PubMed
-
- Forsyth WR, Matthews CR. Folding Mechanism of Indole-3-glycerol Phosphate Synthase from Sulfolobus solfataricus: A Test of the Conservation of Folding Mechanisms Hypothesis in (βα)8 Barrels. J Mol Biol. 2002;320:1119–1133. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
