

AICAR positively regulate glycogen synthase activity and LDL receptor expression through Raf-1/MEK/p42/44MAPK/p90RSK/GSK-3 signaling cascade
- PMID: 17945190
- PMCID: PMC5312663
- DOI: 10.1016/j.bcp.2007.08.028
AICAR positively regulate glycogen synthase activity and LDL receptor expression through Raf-1/MEK/p42/44MAPK/p90RSK/GSK-3 signaling cascade
Abstract
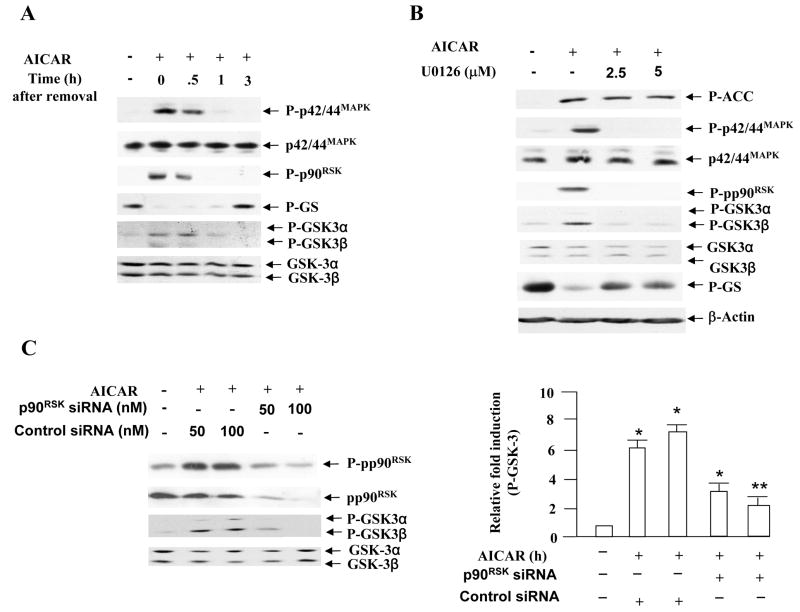
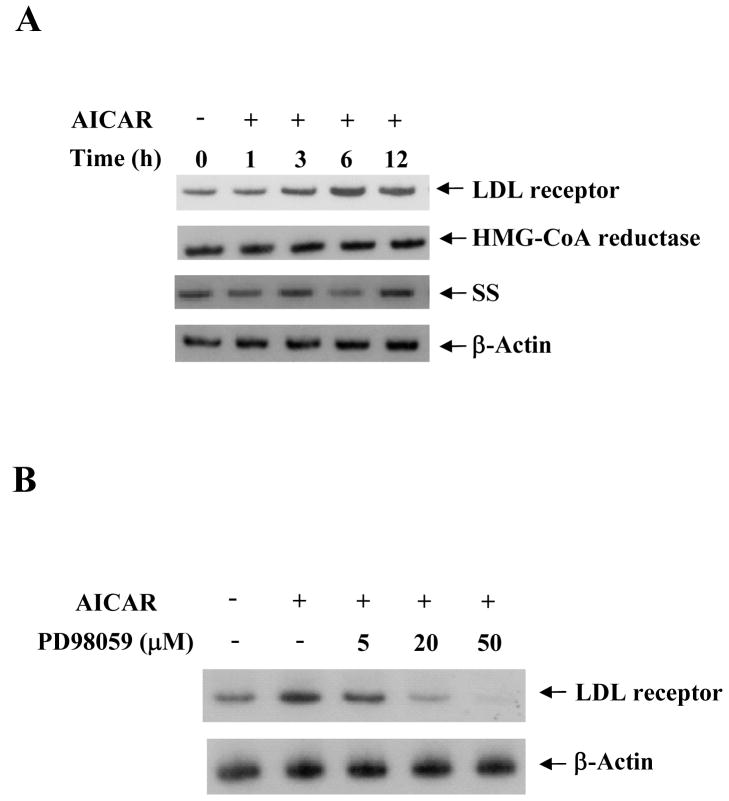
5-Aminoimidazole-4-carboxamide-1-beta-D-ribofuranoside (AICAR) is a commonly used pharmacological agent to study physiological effects which are similar to those of exercise. However, signal transduction pathways by which AICAR elicits downstream effects in liver are poorly understood. We report here that AICAR not only activated AMPK but also phosphorylated/deactivated glycogen synthase kinase-3 alpha/beta (GSK-3alpha/beta) and dephophorylated/activated glycogen synthase (GS) in a time-dependent manner in human hepatoma HepG2 cells. The signal connection between AICAR and GSK-3 is indirect and involves activation of Raf-1/MEK/p42/44(MAPK)/p90(RSK) signaling cascade as pharmacologic inhibition of MEK significantly reduced phosphorylation/deactivation of GSK-3 and consequent dephosphorylation/activation of GS. Moreover, silencing the expression of p90(RSK), a substrate of p42/44(MAPK), attenuated AICAR-dependent GSK-3 phosphorylation, implicating this kinase as a key mediator of AICAR signaling to GSK-3. Furthermore, consistent with the involvement of Raf-1 kinase cascade, AICAR-induced low-density lipoprotein (LDL) receptor expression in a p42/44(MAPK)-dependent manner. Finally, AICAR requires AMPK-alpha2-dependent and -independent pathways to activate Raf-1 kinase cascade as suppression of AMPKalpha2 activity, and not of AMPKalpha1, partially blocked AICAR-dependent p42/44(MAPK) activation and GSK-3 phosphorylation/deactivation. Collectively, these results highlight Raf-1 signaling cascade as the critical mediator of AICAR action on glucose and lipid metabolism in HepG2 cells.
Figures




Similar articles
-
Inhibition of insulin-stimulated glycogen synthesis by 5-aminoimidasole-4-carboxamide-1-beta-d-ribofuranoside-induced adenosine 5'-monophosphate-activated protein kinase activation: interactions with Akt, glycogen synthase kinase 3-3alpha/beta, and glycogen synthase in isolated rat soleus muscle.Endocrinology. 2006 Nov;147(11):5170-7. doi: 10.1210/en.2006-0480. Epub 2006 Jul 27. Endocrinology. 2006. PMID: 16873531
-
MEK inhibitors block AICAR-induced maturation in mouse oocytes by a MAPK-independent mechanism.Mol Reprod Dev. 2005 Feb;70(2):235-45. doi: 10.1002/mrd.20200. Mol Reprod Dev. 2005. PMID: 15570612
-
Insulin signalling downstream of protein kinase B is potentiated by 5'AMP-activated protein kinase in rat hearts in vivo.Diabetologia. 2005 Dec;48(12):2591-601. doi: 10.1007/s00125-005-0016-3. Epub 2005 Nov 11. Diabetologia. 2005. PMID: 16283248
-
Selective suppression of AMP-activated protein kinase in skeletal muscle: update on 'lazy mice'.Biochem Soc Trans. 2003 Feb;31(Pt 1):236-41. doi: 10.1042/bst0310236. Biochem Soc Trans. 2003. PMID: 12546693 Review.
-
GSK-3: tricks of the trade for a multi-tasking kinase.J Cell Sci. 2003 Apr 1;116(Pt 7):1175-86. doi: 10.1242/jcs.00384. J Cell Sci. 2003. PMID: 12615961 Free PMC article. Review.
Cited by
-
AMPK activation by liquiritigenin inhibited oxidative hepatic injury and mitochondrial dysfunction induced by nutrition deprivation as mediated with induction of farnesoid X receptor.Eur J Nutr. 2017 Mar;56(2):635-647. doi: 10.1007/s00394-015-1107-7. Epub 2015 Dec 8. Eur J Nutr. 2017. PMID: 26646674
-
Regulation of ERK1/2 by ouabain and Na-K-ATPase-dependent energy utilization and AMPK activation in parotid acinar cells.Am J Physiol Cell Physiol. 2008 Sep;295(3):C590-9. doi: 10.1152/ajpcell.00140.2008. Epub 2008 Jul 16. Am J Physiol Cell Physiol. 2008. PMID: 18632735 Free PMC article.
-
AMPK-associated signaling to bridge the gap between fuel metabolism and hepatocyte viability.World J Gastroenterol. 2010 Aug 14;16(30):3731-42. doi: 10.3748/wjg.v16.i30.3731. World J Gastroenterol. 2010. PMID: 20698033 Free PMC article. Review.
-
Loss of hepatic AMP-activated protein kinase impedes the rate of glycogenolysis but not gluconeogenic fluxes in exercising mice.J Biol Chem. 2017 Dec 8;292(49):20125-20140. doi: 10.1074/jbc.M117.811547. Epub 2017 Oct 16. J Biol Chem. 2017. PMID: 29038293 Free PMC article.
References
-
- Greenberg CC, Jurczak MJ, Danos AM, Brady MJ. Glycogen branches out: new perspectives on the role of glycogen metabolism in the integration of metabolic pathways. Am J Physiol Endocrinol Metab. 2006;291:E1–E8. - PubMed
-
- Lawrence JC, Roach PJ. New insights into the role and mechanism of glycogen synthase activation by insulin. Diabetes. 1997;46:541–547. - PubMed
-
- Roach PJ. Control of glycogen synthase by hierarchal protein phosphorylation. FASEB J. 1990;4:2961–2968. - PubMed
-
- Parker PJ, Caudwell FB, Cohen P. Glycogen synthase from rabbit skeletal muscle: effect of insulin on the state of phosphorylation of the seven phosphoserine residues in vivo. Eur J Biochem. 1983;130:227–234. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
