

Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors
- PMID: 17947237
- PMCID: PMC3405982
- DOI: 10.1074/jbc.M708137200
Control of phenotypic plasticity of smooth muscle cells by bone morphogenetic protein signaling through the myocardin-related transcription factors
Abstract
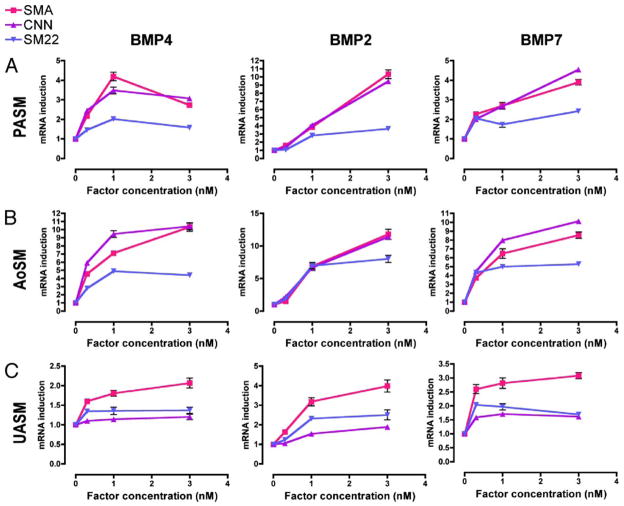
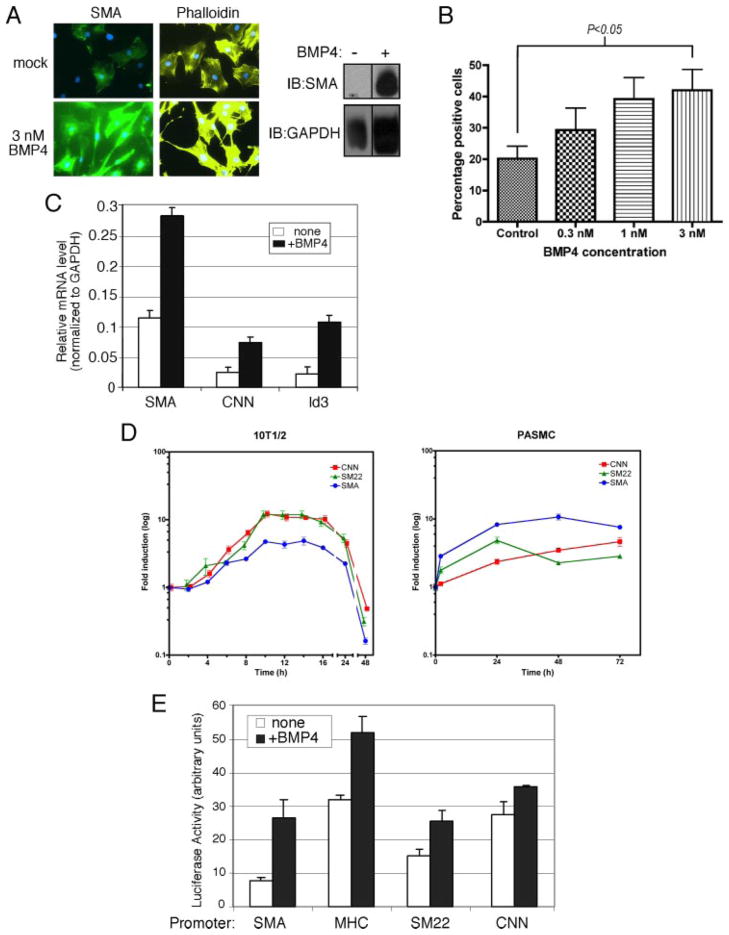
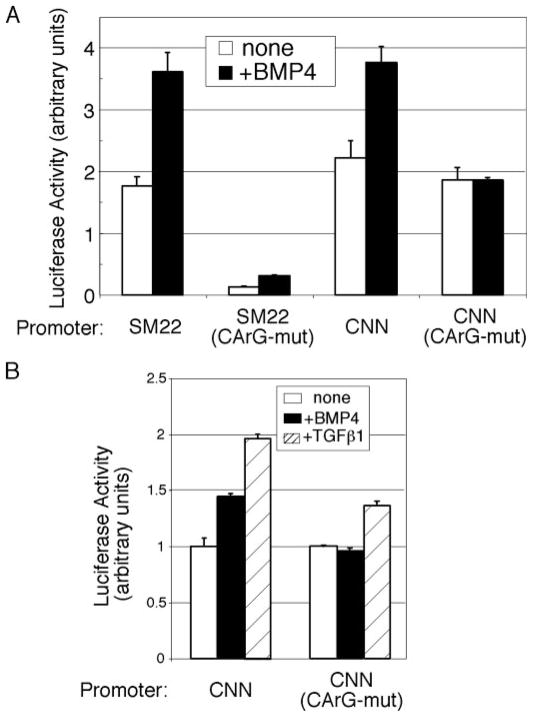
Vascular smooth muscle cells (VSMCs), unlike other muscle cells, do not terminally differentiate. In response to injury, VSMCs change phenotype, proliferate, and migrate as part of the repair process. Dysregulation of this plasticity program contributes to the pathogenesis of several vascular disorders, such as atherosclerosis, restenosis, and hypertension. The discovery of mutations in the gene encoding BMPRII, the type II subunit of the receptor for bone morphogenetic proteins (BMPs), in patients with pulmonary arterial hypertension (PAH) provided an indication that BMP signaling may affect the homeostasis of VSMCs and their phenotype modulation. Here we report that BMP signaling potently induces SMC-specific genes in pluripotent cells and prevents dedifferentiation of arterial SMCs. The BMP-induced phenotype switch requires intact RhoA/ROCK signaling but is not blocked by inhibitors of the TGFbeta and PI3K/Akt pathways. Furthermore, nuclear localization and recruitment of the myocardin-related transcription factors (MRTF-A and MRTF-B) to a smooth muscle alpha-actin promoter is observed in response to BMP treatment. Thus, BMP signaling modulates VSMC phenotype via cross-talk with the RhoA/MRTFs pathway, and may contribute to the development of the pathological characteristics observed in patients with PAH and other obliterative vascular diseases.
Figures




References
-
- Owens G. Physiol Rev. 1995;75:487–517. - PubMed
-
- Rubin LJ, Rich S. Primary Pulmonary Hypertension. Marcel Dekker, Inc; New York, NY: 1997.
-
- Runo JR, Loyd JE. Lancet. 2003;361:1533–1544. - PubMed
-
- Machado RD, Pauciulo MW, Thomson JR, Lane KB, Morgan NV, Wheeler L, Phillips JA, 3rd, Newman J, Williams D, Galie N, Manes A, McNeil K, Yacoub M, Mikhail G, Rogers P, Corris P, Humbert M, Donnai D, Martensson G, Tranebjaerg L, Loyd JE, Trembath RC, Nichols WC. Am J Hum Genet. 2001;68:92–102. - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
