

Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load
- PMID: 17947312
- PMCID: PMC2375504
- DOI: 10.1113/jphysiol.2007.142448
Corticostriatal synaptic function in mouse models of Huntington's disease: early effects of huntingtin repeat length and protein load
Abstract
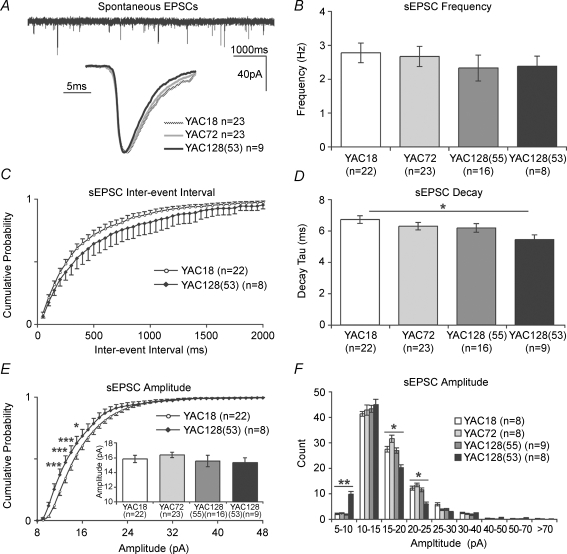
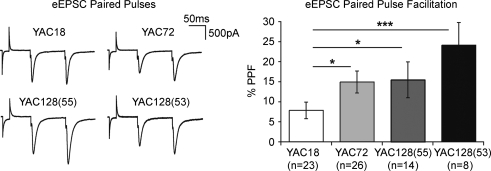
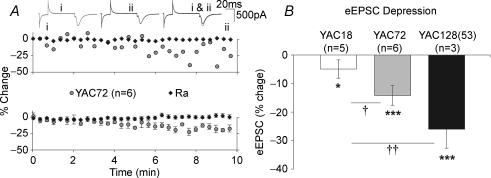
Huntington's disease (HD) is an autosomal dominant, late onset, neurodegenerative disease characterized by motor deficits and dementia that is caused by expansion of a CAG repeat in the HD gene. Clinical manifestations result from selective neuronal degeneration of predominantly GABAergic striatal medium-sized spiny neurons (MSNs). A growing number of studies demonstrate that personality, mood and cognitive disturbances are some of the earliest signs of HD and may reflect synaptic dysfunction prior to neuronal loss. Previous studies in striatal MSNs demonstrated early alterations in NMDA-type glutamate receptor currents in several HD mouse models, as well as evidence for presynaptic dysfunction prior to disease manifestations in the R6/2 HD fragment mouse model. We have compared corticostriatal synaptic function in full-length, human HD gene-carrying YAC transgenic mice expressing a non-pathogenic CAG repeat (YAC18; control) with three increasingly severe variants of pathogenic HD gene-expressing mice (YAC72 and two different lines of YAC128), at ages that precede any detectable disease phenotype. We report presynaptic dysfunction and a propensity towards synaptic depression in YAC72 and YAC128 compared to YAC18 mice, and, in the most severe model, we also observed altered AMPA receptor function. When normalized to evoked AMPAR currents, postsynaptic NMDAR currents are augmented in all three pathogenic HD YAC variants. These findings demonstrate multiple perturbations to corticostriatal synaptic function in HD mice, furthering our understanding of the early effects of the HD mutation that may contribute to cognitive dysfunction, mood disorders and later development of more serious dysfunction. Furthermore, this study provides a set of neurophysiological sequelae against which to test and compare other mouse models and potential therapies in HD.
Figures

Similar articles
-
Enhanced striatal NR2B-containing N-methyl-D-aspartate receptor-mediated synaptic currents in a mouse model of Huntington disease.J Neurophysiol. 2004 Nov;92(5):2738-46. doi: 10.1152/jn.00308.2004. Epub 2004 Jul 7. J Neurophysiol. 2004. PMID: 15240759
-
Differential changes in thalamic and cortical excitatory synapses onto striatal spiny projection neurons in a Huntington disease mouse model.Neurobiol Dis. 2016 Feb;86:62-74. doi: 10.1016/j.nbd.2015.11.020. Epub 2015 Nov 24. Neurobiol Dis. 2016. PMID: 26621114
-
Role of NR2B-type NMDA receptors in selective neurodegeneration in Huntington disease.Neurobiol Aging. 2003 Dec;24(8):1113-21. doi: 10.1016/j.neurobiolaging.2003.04.003. Neurobiol Aging. 2003. PMID: 14643383
-
Pathophysiology of Huntington's disease: time-dependent alterations in synaptic and receptor function.Neuroscience. 2011 Dec 15;198:252-73. doi: 10.1016/j.neuroscience.2011.08.052. Epub 2011 Aug 27. Neuroscience. 2011. PMID: 21907762 Free PMC article. Review.
-
Functional interactions within striatal microcircuit in animal models of Huntington's disease.Neuroscience. 2012 Jun 1;211:165-84. doi: 10.1016/j.neuroscience.2011.06.075. Epub 2011 Jul 1. Neuroscience. 2012. PMID: 21756979 Review.
Cited by
-
C57BL/6 Background Attenuates mHTT Toxicity in the Striatum of YAC128 Mice.Int J Mol Sci. 2021 Nov 23;22(23):12664. doi: 10.3390/ijms222312664. Int J Mol Sci. 2021. PMID: 34884469 Free PMC article.
-
Microglia and complement mediate early corticostriatal synapse loss and cognitive dysfunction in Huntington's disease.Nat Med. 2023 Nov;29(11):2866-2884. doi: 10.1038/s41591-023-02566-3. Epub 2023 Oct 9. Nat Med. 2023. PMID: 37814059 Free PMC article.
-
Inflammation alters AMPA-stimulated calcium responses in dorsal striatal D2 but not D1 spiny projection neurons.Eur J Neurosci. 2017 Nov;46(9):2519-2533. doi: 10.1111/ejn.13711. Epub 2017 Oct 10. Eur J Neurosci. 2017. PMID: 28921719 Free PMC article.
-
Ca(2+) handling in isolated brain mitochondria and cultured neurons derived from the YAC128 mouse model of Huntington's disease.J Neurochem. 2015 Aug;134(4):652-67. doi: 10.1111/jnc.13165. Epub 2015 Jun 4. J Neurochem. 2015. PMID: 25963273 Free PMC article.
-
Altered Regulation of Striatal Neuronal N-Methyl-D-Aspartate Receptor Trafficking by Palmitoylation in Huntington Disease Mouse Model.Front Synaptic Neurosci. 2019 Feb 21;11:3. doi: 10.3389/fnsyn.2019.00003. eCollection 2019. Front Synaptic Neurosci. 2019. PMID: 30846936 Free PMC article.
References
-
- Andre VM, Cepeda C, Venegas A, Gomez Y, Levine MS. Altered cortical glutamate receptor function in the R6/2 model of Huntington's disease. J Neurophysiol. 2006;95:2108–2119. - PubMed
-
- Aronin N, Chase K, Young C, Sapp E, Schwarz C, Matta N, Kornreich R, Landwehrmeyer B, Bird E, Beal MF, et al. CAG expansion affects the expression of mutant Huntingtin in the Huntington's disease brain. Neuron. 1995;15:1193–1201. - PubMed
-
- Beal MF. Role of excitotoxicity in human neurological disease. Curr Opin Neurobiol. 1992;2:657–662. - PubMed
-
- Berrios GE, Wagle AC, Marková IS, Wagle SA, Rosser A, Hodges JR. Psychiatric symptoms in neurologically asymptomatic Huntington's disease gene carriers: a comparison with gene negative at risk subjects. Acta Psychiatr Scand. 2002;105:224–230. - PubMed
-
- Bliss TVP, Collingridge GL. A synaptic model of memory: long-term potentiation in the hippocampus. Nature. 1993;361:31–39. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases