

Designing candidate gene and genome-wide case-control association studies
- PMID: 17947991
- PMCID: PMC4180089
- DOI: 10.1038/nprot.2007.366
Designing candidate gene and genome-wide case-control association studies
Abstract
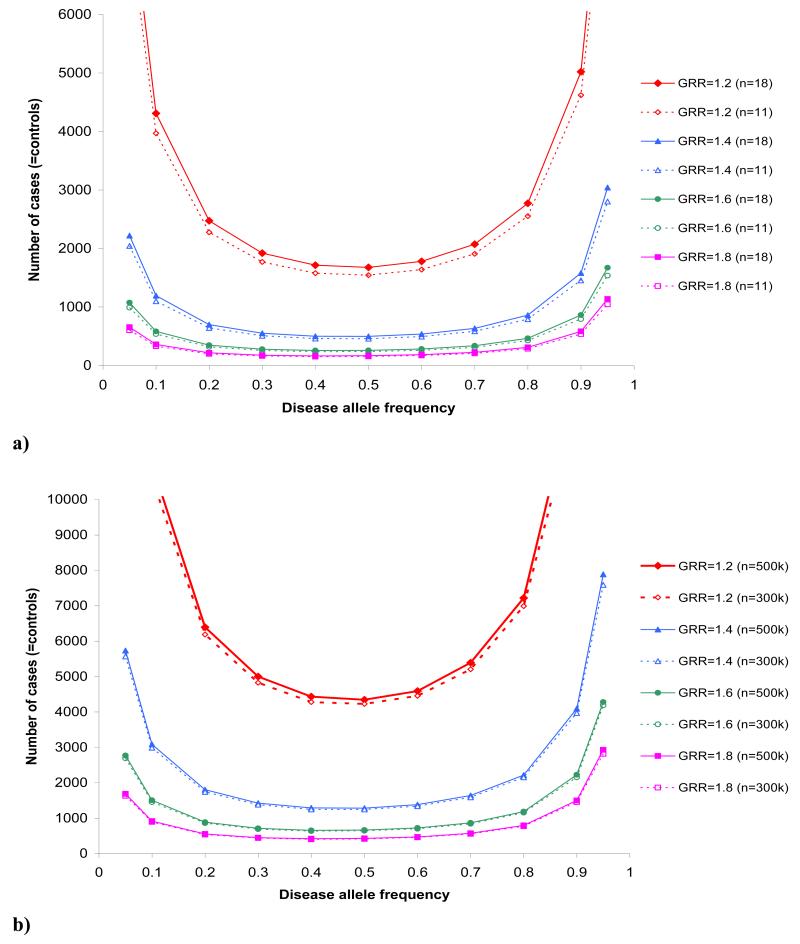
This protocol describes how to appropriately design a genetic association case-control study, either focusing on a candidate gene (CG) or region or implementing a genome-wide approach. The steps described involve: (i) defining the case phenotype in adequate detail; (ii) checking the heritability of the disease in question; (iii) considering whether a population-based study is the appropriate design for the research question; (iv) the appropriate selection of controls; (v) sample size calculations and (vi) giving due consideration to whether it is a de novo or replication study. General guidelines are given, as well as specific examples of a CG and a genome-wide association study into type 2 diabetes. Software and websites used in this protocol include the International HapMap Consortium website, Genetic Power Calculator, CaT, and SNPSpD. Running each of the programs takes only a few seconds; the rate-limiting steps involve thinking through the designs and parameters in the disease models.
Figures
References
-
- Gilliam TC, et al. Localization of the Huntington’s disease gene to a small segment of chromosome 4 flanked by D4S10 and the telomere. Cell. 1987;50:565–71. - PubMed
-
- Kerem B, et al. Identification of the cystic fibrosis gene: genetic analysis. Science. 1989;245:1073–80. - PubMed
-
- Palmer LJ, Cardon LR. Shaking the tree: mapping complex disease genes with linkage disequilibrium. Lancet. 2005;366:1223–34. - PubMed
-
- Zondervan KT, Cardon LR. The complex interplay among factors that influence allelic association. Nat Rev Genet. 2004;5:89–100. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
