

DMS footprinting of structured RNAs and RNA-protein complexes
- PMID: 17948004
- PMCID: PMC2701642
- DOI: 10.1038/nprot.2007.380
DMS footprinting of structured RNAs and RNA-protein complexes
Abstract
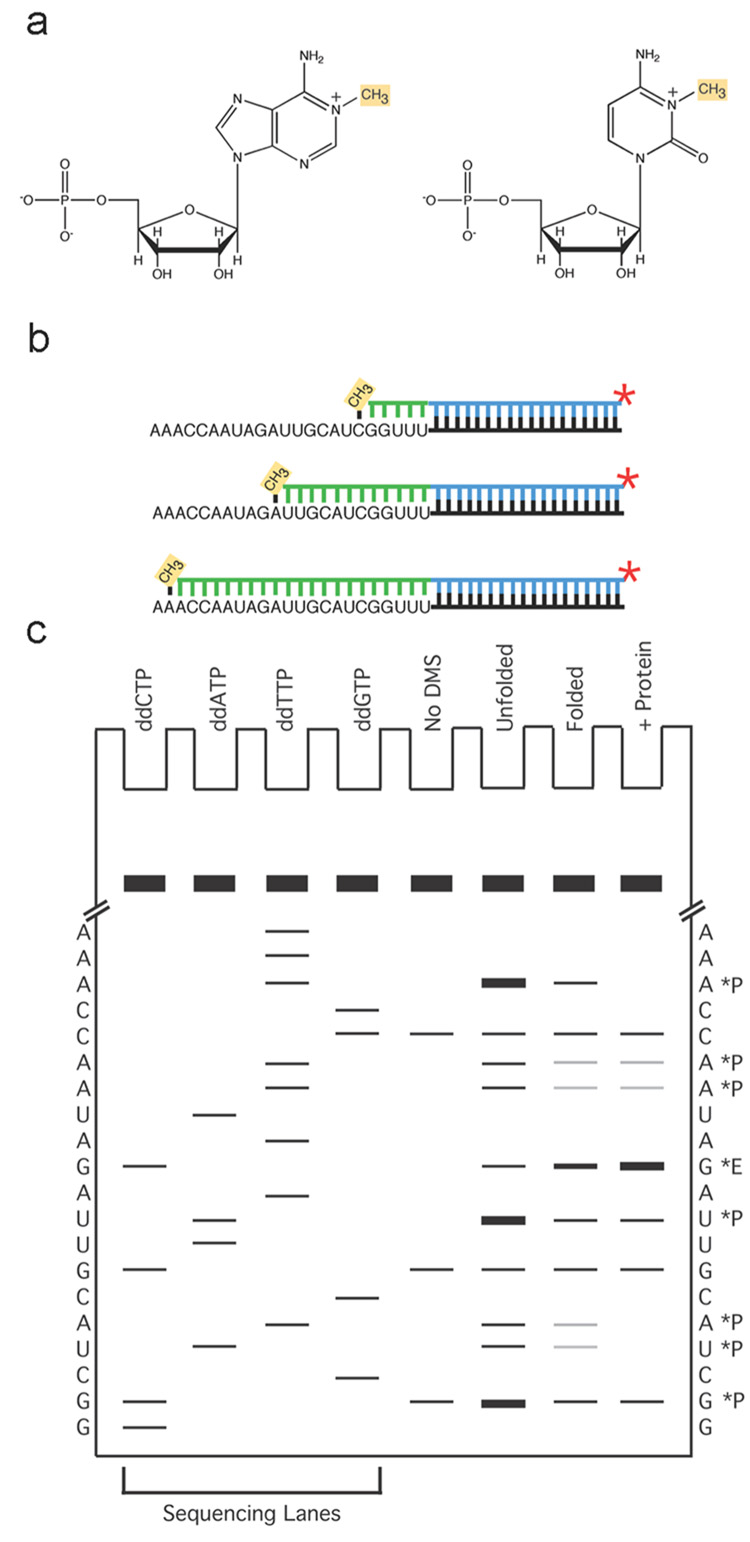
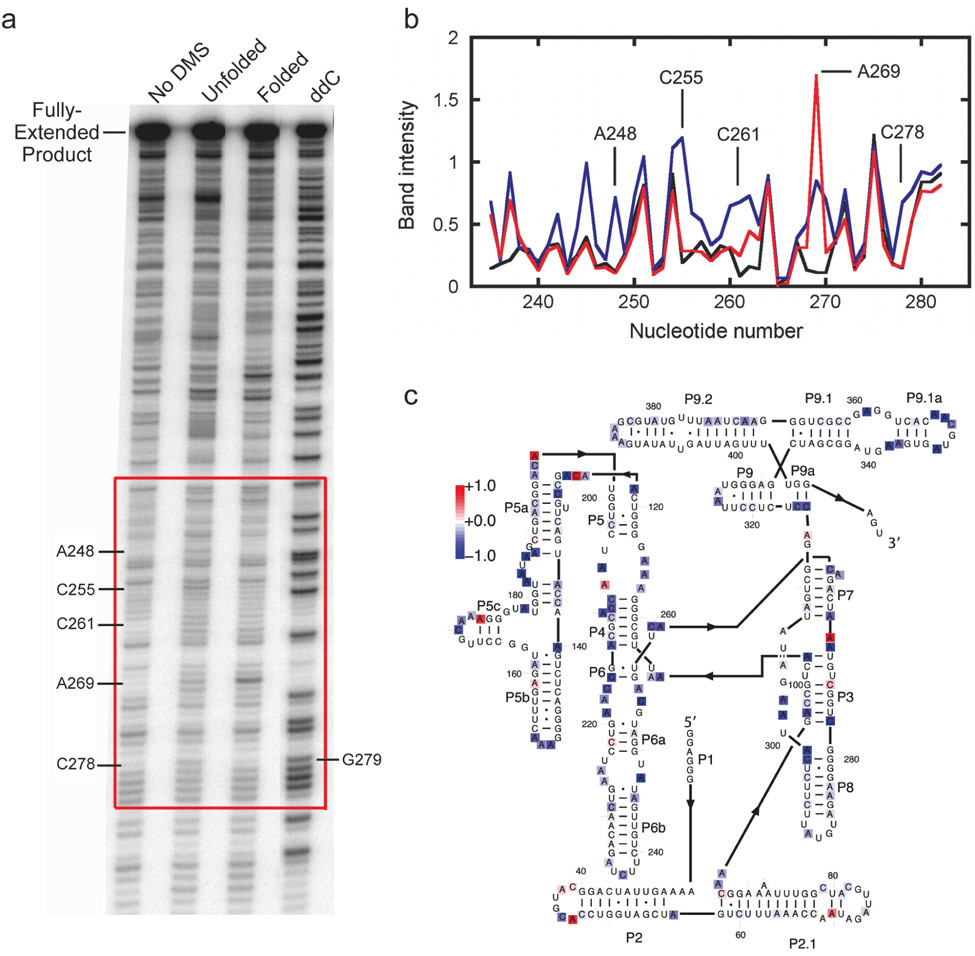
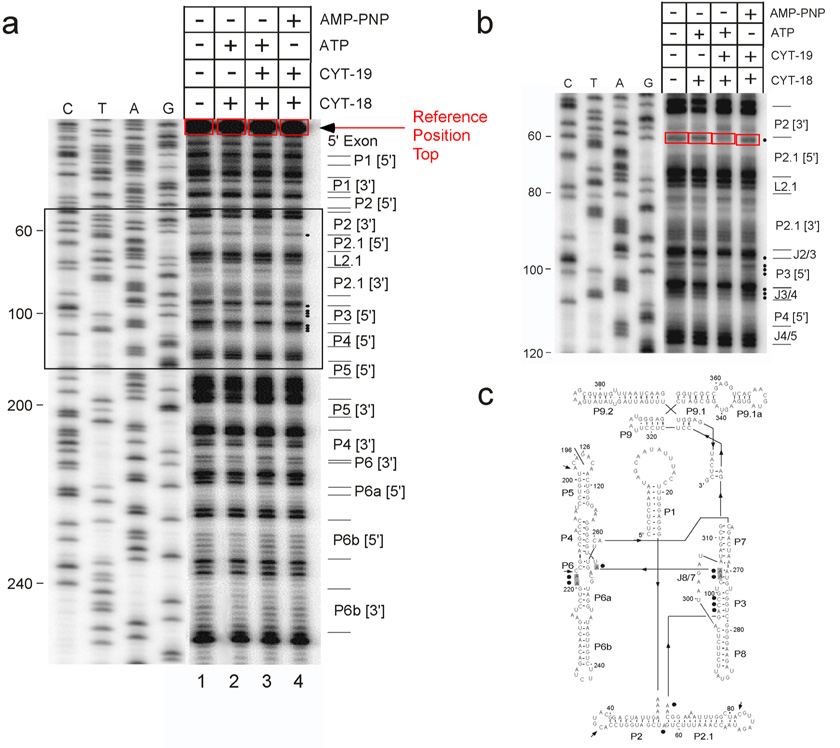
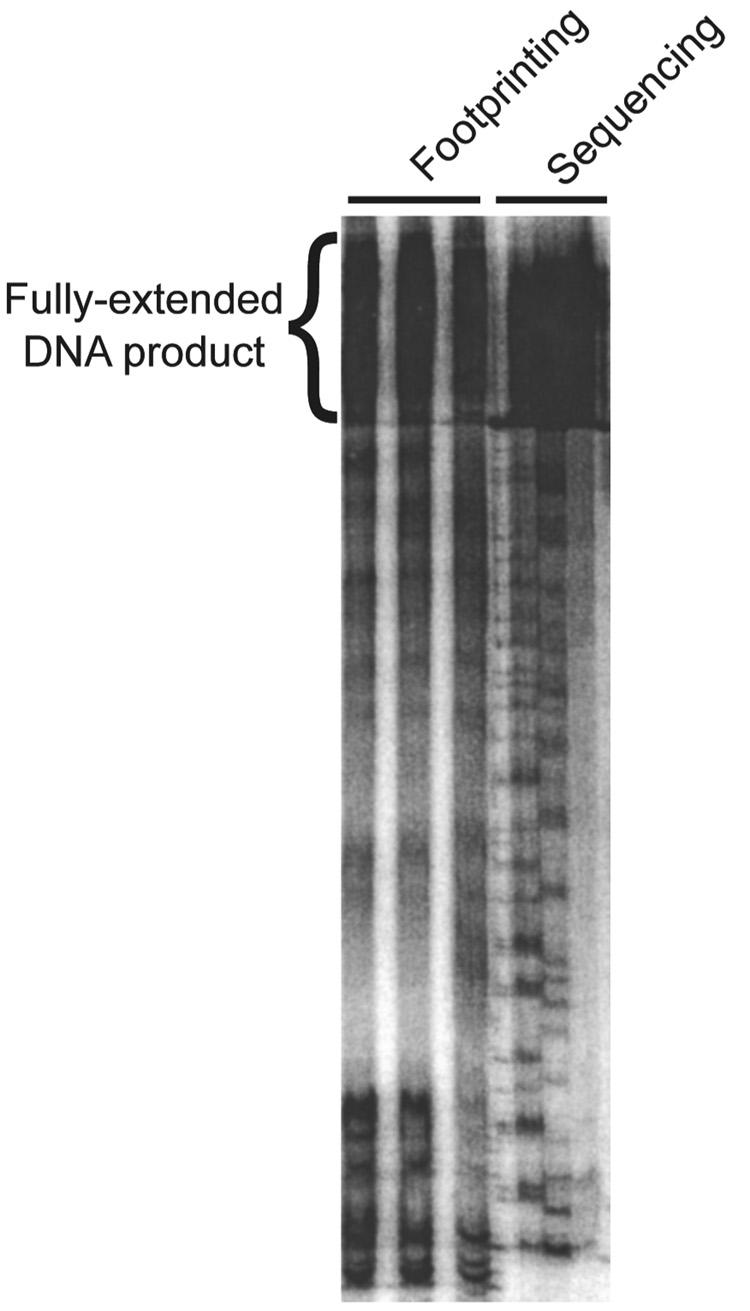
We describe a protocol in which dimethyl sulfate (DMS) modification of the base-pairing faces of unpaired adenosine and cytidine nucleotides is used for structural analysis of RNAs and RNA-protein complexes (RNPs). The protocol is optimized for RNAs of small to moderate size (< or = 500 nt). The RNA or RNP is first exposed to DMS under conditions that promote formation of the folded structure or complex, as well as 'control' conditions that do not allow folding or complex formation. The positions and extents of modification are then determined by primer extension, polyacrylamide gel electrophoresis and quantitative analysis. From changes in the extent of modification upon folding or protein binding (appearance of a 'footprint'), it is possible to detect local changes in the secondary and tertiary structure of RNA, as well as the formation of RNA-protein contacts. This protocol takes 1.5-3 d to complete, depending on the type of analysis used.
Figures

References
-
- Golden BL. Preparation and crystallization of RNA. Methods Mol. Biol. 363;2007:239–257. - PubMed
-
- Wu H, Finger LD, Feigon J. Structure determination of protein/RNA complexes by NMR. Methods Enzymol. 2005;394:525–545. - PubMed
-
- Myong S, Stevens BC, Ha T. Bridging conformational dynamics and function using single-molecule spectroscopy. Structure. 2006;14:633–643. - PubMed
-
- Lipfert J, Doniach S. Small-angle X-ray scattering from RNA, proteins, and protein complexes. Annu. Rev. Biophys. Biomol. Struct. 2007;36:307–327. - PubMed
-
- Kim NK, Murali A, DeRose VJ. A distance ruler for RNA using EPR and site-directed spin labeling. Chem. Biol. 2004;11:939–948. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
