

Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation?
- PMID: 17948061
- PMCID: PMC2080798
- DOI: 10.1038/sj.emboj.7601885
Spinocerebellar ataxia with axonal neuropathy: consequence of a Tdp1 recessive neomorphic mutation?
Abstract
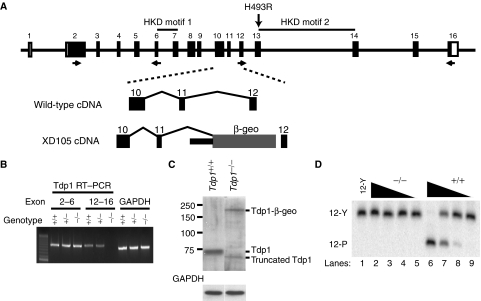
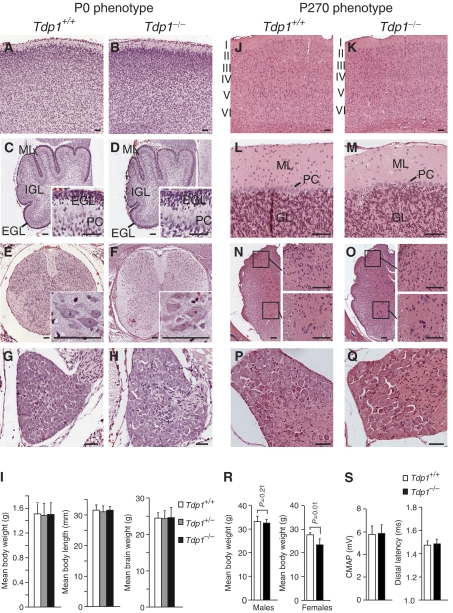
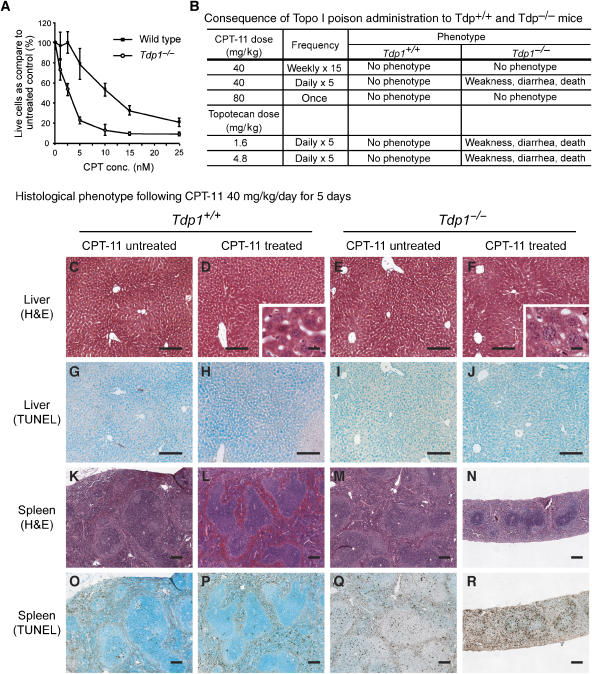
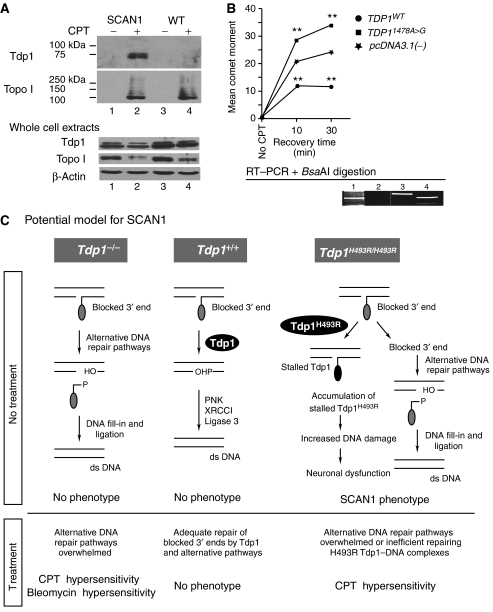
Tyrosyl-DNA phosphodiesterase 1 (Tdp1) cleaves the phosphodiester bond between a covalently stalled topoisomerase I (Topo I) and the 3' end of DNA. Stalling of Topo I at DNA strand breaks is induced by endogenous DNA damage and the Topo I-specific anticancer drug camptothecin (CPT). The H493R mutation of Tdp1 causes the neurodegenerative disorder spinocerebellar ataxia with axonal neuropathy (SCAN1). Contrary to the hypothesis that SCAN1 arises from catalytically inactive Tdp1, Tdp1-/- mice are indistinguishable from wild-type mice, physically, histologically, behaviorally, and electrophysiologically. However, compared to wild-type mice, Tdp1-/- mice are hypersensitive to CPT and bleomycin but not to etoposide. Consistent with earlier in vitro studies, we show that the H493R Tdp1 mutant protein retains residual activity and becomes covalently trapped on the DNA after CPT treatment of SCAN1 cells. This result provides a direct demonstration that Tdp1 repairs Topo I covalent lesions in vivo and suggests that SCAN1 arises from the recessive neomorphic mutation H493R. This is a novel mechanism for disease since neomorphic mutations are generally dominant.
Figures


Similar articles
-
In vitro complementation of Tdp1 deficiency indicates a stabilized enzyme-DNA adduct from tyrosyl but not glycolate lesions as a consequence of the SCAN1 mutation.DNA Repair (Amst). 2009 May 1;8(5):654-63. doi: 10.1016/j.dnarep.2008.12.012. Epub 2009 Feb 10. DNA Repair (Amst). 2009. PMID: 19211312 Free PMC article.
-
SCAN1 mutant Tdp1 accumulates the enzyme--DNA intermediate and causes camptothecin hypersensitivity.EMBO J. 2005 Jun 15;24(12):2224-33. doi: 10.1038/sj.emboj.7600694. Epub 2005 May 26. EMBO J. 2005. PMID: 15920477 Free PMC article.
-
Hereditary ataxia SCAN1 cells are defective for the repair of transcription-dependent topoisomerase I cleavage complexes.DNA Repair (Amst). 2006 Dec 9;5(12):1489-94. doi: 10.1016/j.dnarep.2006.07.004. Epub 2006 Aug 28. DNA Repair (Amst). 2006. PMID: 16935573
-
Spinocerebellar ataxia with axonal neuropathy.Adv Exp Med Biol. 2010;685:75-83. doi: 10.1007/978-1-4419-6448-9_7. Adv Exp Med Biol. 2010. PMID: 20687496 Review.
-
DNA single-strand break repair and spinocerebellar ataxia with axonal neuropathy-1.Neuroscience. 2007 Apr 14;145(4):1260-6. doi: 10.1016/j.neuroscience.2006.08.048. Epub 2006 Oct 11. Neuroscience. 2007. PMID: 17045754 Review.
Cited by
-
The Interaction of the Metallo-Glycopeptide Anti-Tumour Drug Bleomycin with DNA.Int J Mol Sci. 2018 May 4;19(5):1372. doi: 10.3390/ijms19051372. Int J Mol Sci. 2018. PMID: 29734689 Free PMC article. Review.
-
DNA Damage Response and Repair, DNA Methylation, and Cell Death in Human Neurons and Experimental Animal Neurons Are Different.J Neuropathol Exp Neurol. 2018 Jul 1;77(7):636-655. doi: 10.1093/jnen/nly040. J Neuropathol Exp Neurol. 2018. PMID: 29788379 Free PMC article.
-
A Lysine Desert Protects a Novel Domain in the Slx5-Slx8 SUMO Targeted Ub Ligase To Maintain Sumoylation Levels in Saccharomyces cerevisiae.Genetics. 2017 Aug;206(4):1807-1821. doi: 10.1534/genetics.117.202697. Epub 2017 May 26. Genetics. 2017. PMID: 28550017 Free PMC article.
-
DNA repair deficiency and neurological disease.Nat Rev Neurosci. 2009 Feb;10(2):100-12. doi: 10.1038/nrn2559. Epub 2009 Jan 15. Nat Rev Neurosci. 2009. PMID: 19145234 Free PMC article. Review.
-
Synergistic decrease of DNA single-strand break repair rates in mouse neural cells lacking both Tdp1 and aprataxin.DNA Repair (Amst). 2009 Jun 4;8(6):760-6. doi: 10.1016/j.dnarep.2009.02.002. Epub 2009 Mar 19. DNA Repair (Amst). 2009. PMID: 19303373 Free PMC article.
References
-
- Antonarakis SE, Krawczak M, Cooper DN (2001) The nature and mechanisms of human gene mutation. In The Metabolic & Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds), pp 343–377. New York: McGraw-Hill
-
- Arvidson B (1979) Distribution of intravenously injected protein tracers in peripheral ganglia of adult mice. Exp Neurol 63: 388–410 - PubMed
-
- Barthelmes HU, Habermeyer M, Christensen MO, Mielke C, Interthal H, Pouliot JJ, Boege F, Marko D (2004) TDP1 overexpression in human cells counteracts DNA damage mediated by topoisomerases I and II. J Biol Chem 279: 55618–55625 - PubMed
-
- Beaudet AL, Scriver CR, Sly WS, Valle D (2001) Genetics, biochemistry, and molecular bases of variant human phenotypes. In The Metabolic & Molecular Bases of Inherited Disease, Scriver CR, Beaudet AL, Sly WS, Valle D, Childs B, Kinzler KW, Vogelstein B (eds), pp 3–45. New York: McGraw-Hill
