

Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as "continuum" phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain
- PMID: 17961929
- PMCID: PMC3947608
- DOI: 10.1016/j.neuroscience.2007.06.060
Failure to complete apoptosis following neonatal hypoxia-ischemia manifests as "continuum" phenotype of cell death and occurs with multiple manifestations of mitochondrial dysfunction in rodent forebrain
Abstract
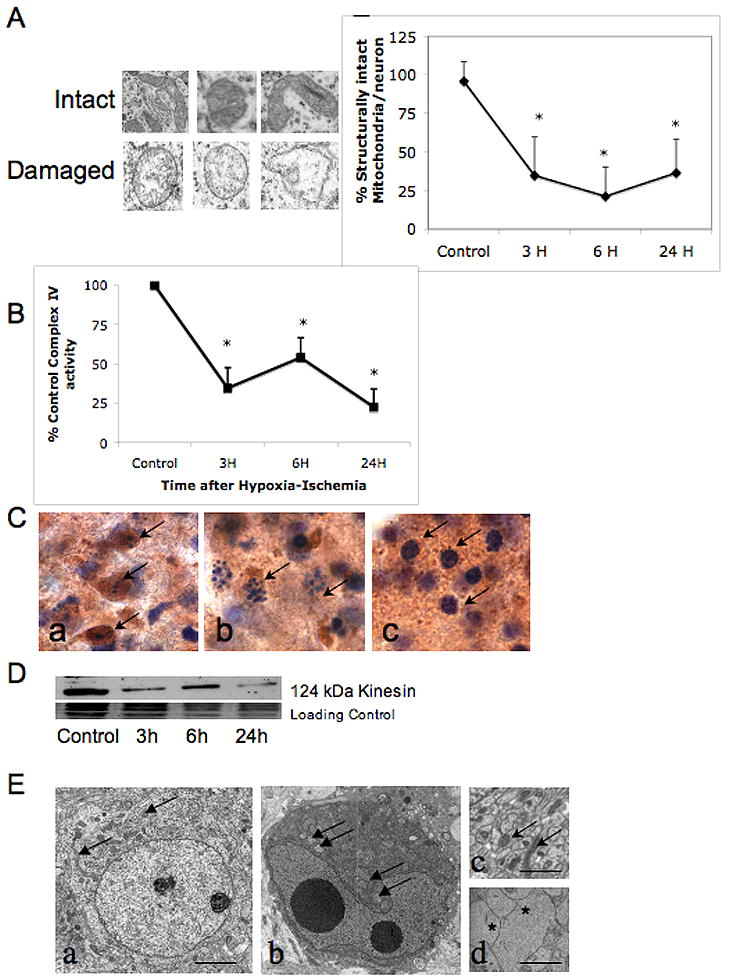
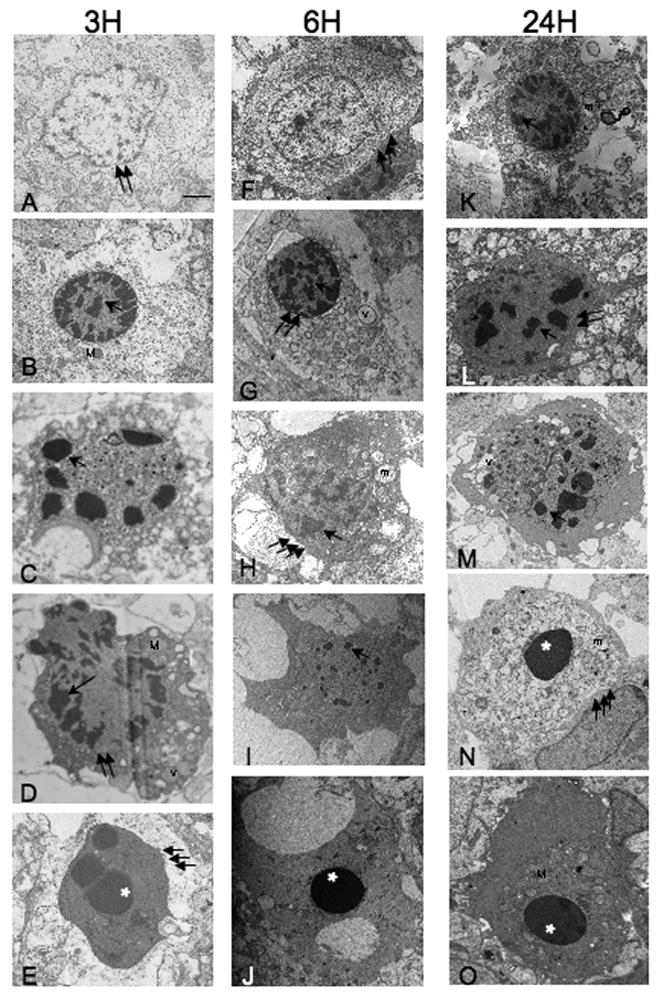
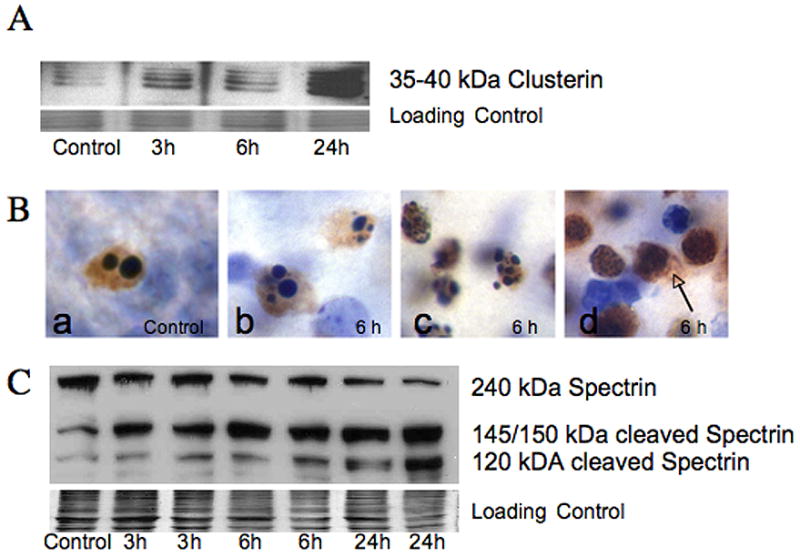
Controversy surrounds proper classification of neurodegeneration occurring acutely following neonatal hypoxia-ischemia (HI). By ultrastructural classification, in the first 24 h after neonatal hypoxia-ischemia in the 7-day-old (p7) rat, the majority of striatal cells die having both apoptotic and necrotic features. There is formation of a functional apoptosome, and activation of caspases-9 and -3 occurring simultaneously with loss of structurally intact mitochondria to 34.7+/-25% and loss of mitochondrial cytochrome c oxidase activity to 34.7+/-12.7% of control levels by 3 h after hypoxia-ischemia. There is also loss of the mitochondrial motor protein, kinesin. This combination of activation of apoptosis pathways simultaneous with significant mitochondrial dysfunction may cause incomplete packaging of nuclear and cytoplasmic contents and a hybrid of necrotic and apoptotic features. Evidence for an intermediate biochemistry of cell death including expression of the 17 kDa isoform of caspase-3 in dying neurons lacking a classic apoptotic morphology and degradation of the neuronal cytoskeletal protein spectrin by caspase-3 and calcium-activated calpains yielding 120 kDa and 145/150 kDa fragments, respectively, is also found. In summary, neonatal hypoxia-ischemia triggers apoptotic cascades, and simultaneously causes mitochondrial structural and functional failure. The presence of a "continuum" phenotype of cell death that varies on a cell-by-cell basis suggests that the phenotype of cell death is dependent on the energy available to drive the apoptotic pathways to completion.
Figures

References
-
- Ankarcrona M, Dypbukt JM, Bonfoco E, Zhivotovsky B, Orrenius S, Lipton SA, Nicotera P. Glutamate-induced neuronal death: a succession of necrosis or apoptosis depending on mitochondrial function. Neuron. 1995;15:961–973. - PubMed
-
- Blomgren K, Hagberg H. Free radicals, mitochondria, and hypoxia-ischemia in the developing brain. Free Radic Biol Med. 2006;40:388–397. - PubMed
-
- Blomgren K, Leist M, Groc L. Pathological apoptosis in the developing brain. Apoptosis. 2007 in press. - PubMed
-
- Blomgren K, Zhu C, Wang X, Karlsson JO, Leverin AL, Bahr BA, Mallard C, Hagberg H. Synergistic activation of caspase-3 by m-calpain after neonatal hypoxia- ischemia: a mechanism of “pathological apoptosis”? J Biol Chem. 2001;276:10191–10198. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
