

A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease
- PMID: 17962415
- PMCID: PMC2077047
- DOI: 10.1073/pnas.0704975104
A common mechanism of PLP/DM20 misfolding causes cysteine-mediated endoplasmic reticulum retention in oligodendrocytes and Pelizaeus-Merzbacher disease
Abstract
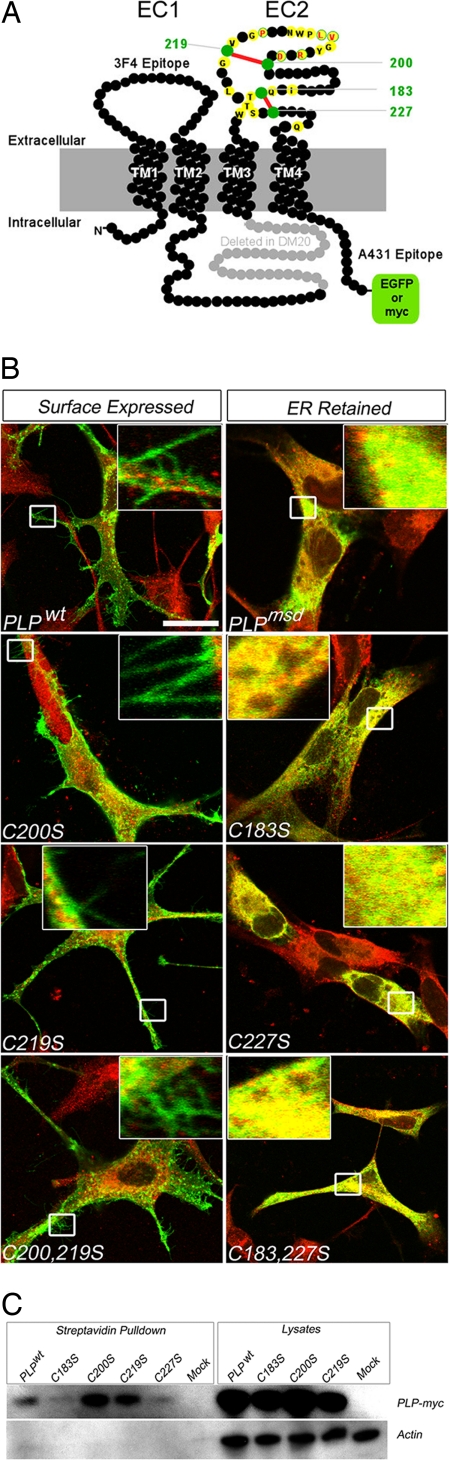
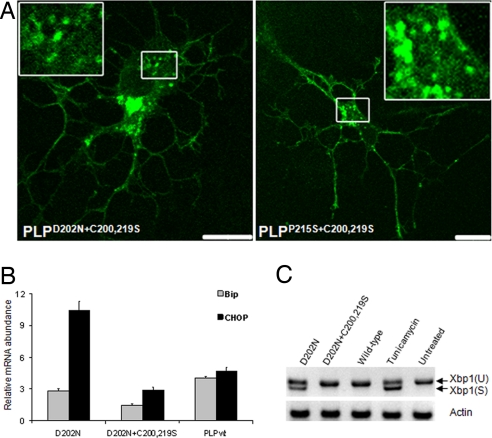
A large number of mutations in the human PLP1 gene lead to abnormal myelination and oligodendrocyte death in Pelizaeus-Merzbacher disease (PMD). Here we show that a major subgroup of PMD mutations that map into the extracellular loop region of PLP/DM20 leads to the failure of oligodendrocytes to form the correct intramolecular disulfide bridges. This leads to abnormal protein cross-links and endoplasmic reticulum retention and activates the unfolded protein response. Importantly, surface expression of mutant PLP/DM20 can be restored and the unfolded protein response can be reverted by the removal of two cysteines. Thus, covalent protein cross-links emerge as a cause, rather than as a consequence, of endoplasmic reticulum retention.
Conflict of interest statement
The authors declare no conflict of interest.
Figures


References
-
- Hurtley SM, Helenius A. Annu Rev Cell Biol. 1989;5:277–307. - PubMed
-
- Ellgaard L, Helenius A. Nat Rev Mol Cell Biol. 2003;4:181–191. - PubMed
-
- Ellgaard L, Molinari M, Helenius A. Science. 1999;286:1882–1888. - PubMed
-
- Rutkowski DT, Kaufman RJ. Trends Cell Biol. 2004;14:20–28. - PubMed
-
- Patil C, Walter P. Curr Opin Cell Biol. 2001;13:349–355. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
