

Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants
- PMID: 17963481
- PMCID: PMC2204015
- DOI: 10.1186/1471-2164-8-391
Database of Trypanosoma cruzi repeated genes: 20,000 additional gene variants
Abstract
Background: Repeats are present in all genomes, and often have important functions. However, in large genome sequencing projects, many repetitive regions remain uncharacterized. The genome of the protozoan parasite Trypanosoma cruzi consists of more than 50% repeats. These repeats include surface molecule genes, and several other gene families. In the T. cruzi genome sequencing project, it was clear that not all copies of repetitive genes were present in the assembly, due to collapse of nearly identical repeats. However, at the time of publication of the T. cruzi genome, it was not clear to what extent this had occurred.
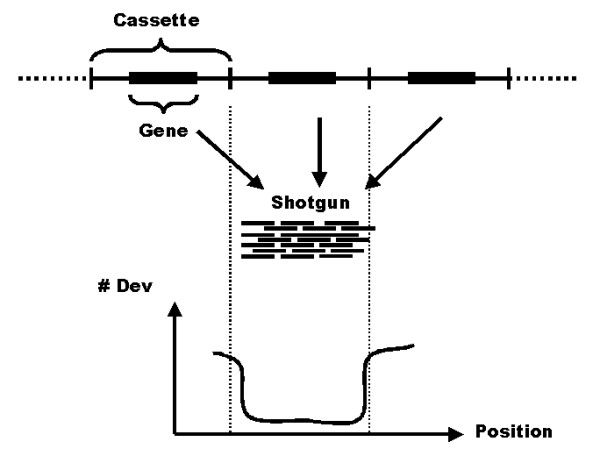
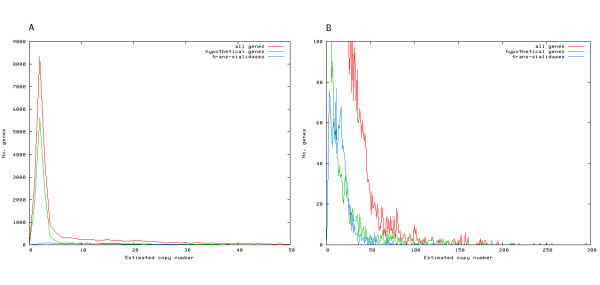
Results: We have developed a pipeline to estimate the genomic repeat content, where shotgun reads are aligned to the genomic sequence and the gene copy number is estimated using the average shotgun coverage. This method was applied to the genome of T. cruzi and copy numbers of all protein coding sequences and pseudogenes were estimated. The 22,640 results were stored in a database available online. 18% of all protein coding sequences and pseudogenes were estimated to exist in 14 or more copies in the T. cruzi CL Brener genome. The average coverage of the annotated protein coding sequences and pseudogenes indicate a total gene copy number, including allelic gene variants, of over 40,000.
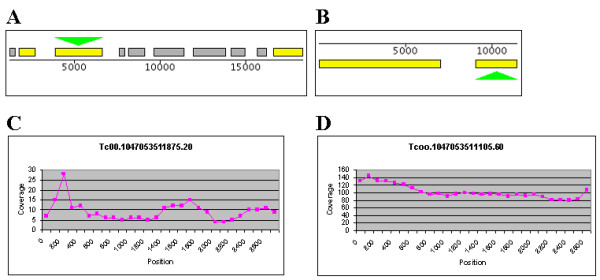
Conclusion: Our results indicate that the number of protein coding sequences and pseudogenes in the T. cruzi genome may be twice the previous estimate. We have constructed a database of the T. cruzi gene repeat data that is available as a resource to the community. The main purpose of the database is to enable biologists interested in repeated, unfinished regions to closely examine and resolve these regions themselves using all available shotgun data, instead of having to rely on annotated consensus sequences that often are erroneous and possibly misleading. Five repetitive genes were studied in more detail, in order to illustrate how the database can be used to analyze and extract information about gene repeats with different characteristics in Trypanosoma cruzi.
Figures

References
-
- Bussey KJ, Chin K, Lababidi S, Reimers M, Reinhold WC, Kuo WL, Gwadry F, Kouros-Mehr H, Fridlyand J, Jain A, Collins C, Nishizuka S, Tonon G, Roschke A, Gehlhaus K, Kirsch I, Scudiero DA, Gray JW, Weinstein JN. Integrating data on DNA copy number with gene expression levels and drug sensitivities in the NCI-60 cell line panel. Mol Cancer Ther. 2006;5:853–867. doi: 10.1158/1535-7163.MCT-05-0155. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
