

How accurately is ncRNA aligned within whole-genome multiple alignments?
- PMID: 17963514
- PMCID: PMC2206062
- DOI: 10.1186/1471-2105-8-417
How accurately is ncRNA aligned within whole-genome multiple alignments?
Abstract
Background: Multiple alignment of homologous DNA sequences is of great interest to biologists since it provides a window into evolutionary processes. At present, the accuracy of whole-genome multiple alignments, particularly in noncoding regions, has not been thoroughly evaluated.
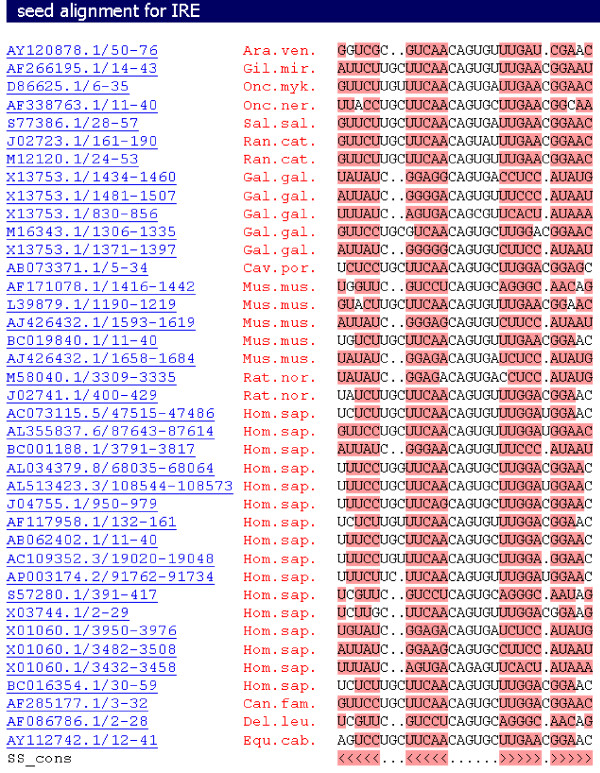
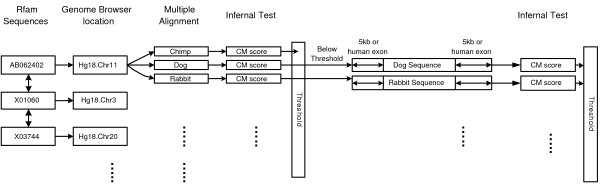
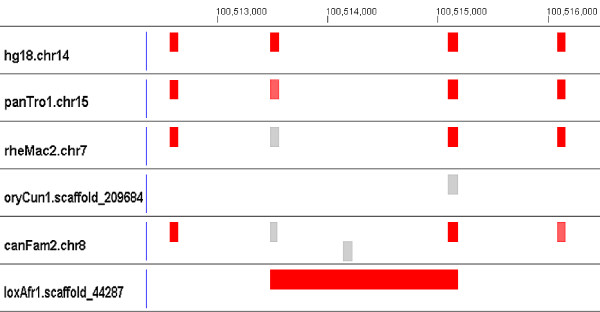
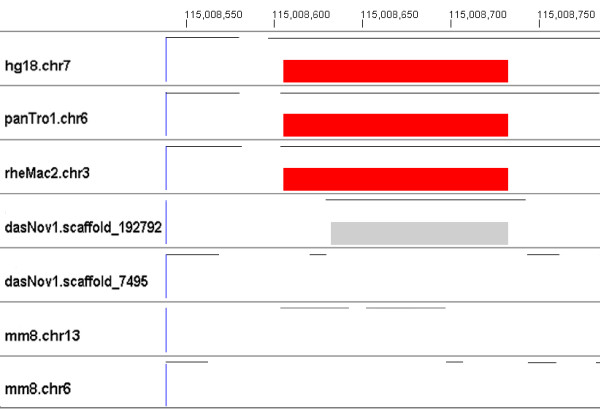
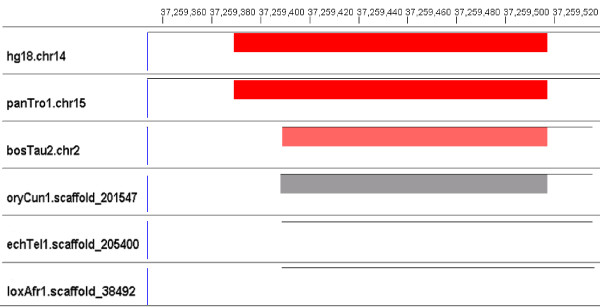
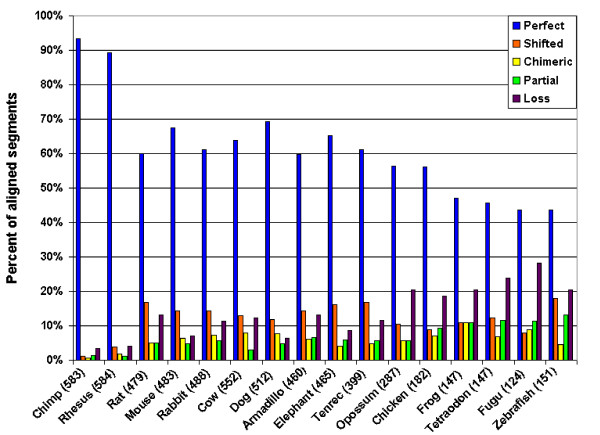
Results: We evaluate the alignment accuracy of certain noncoding regions using noncoding RNA alignments from Rfam as a reference. We inspect the MULTIZ 17-vertebrate alignment from the UCSC Genome Browser for all the human sequences in the Rfam seed alignments. In particular, we find 638 instances of chimeric and partial alignments to human noncoding RNA elements, of which at least 225 can be improved by straightforward means. As a byproduct of our procedure, we predict many novel instances of known ncRNA families that are suggested by the alignment.
Conclusion: MULTIZ does a fairly accurate job of aligning these genomes in these difficult regions. However, our experiments indicate that better alignments exist in some regions.
Figures

Similar articles
-
Multiple whole-genome alignments without a reference organism.Genome Res. 2009 Apr;19(4):682-9. doi: 10.1101/gr.081778.108. Epub 2009 Jan 28. Genome Res. 2009. PMID: 19176791 Free PMC article.
-
Aligning multiple genomic sequences with the threaded blockset aligner.Genome Res. 2004 Apr;14(4):708-15. doi: 10.1101/gr.1933104. Genome Res. 2004. PMID: 15060014 Free PMC article.
-
Inferring noncoding RNA families and classes by means of genome-scale structure-based clustering.PLoS Comput Biol. 2007 Apr 13;3(4):e65. doi: 10.1371/journal.pcbi.0030065. Epub 2007 Feb 22. PLoS Comput Biol. 2007. PMID: 17432929 Free PMC article.
-
Detecting and comparing non-coding RNAs in the high-throughput era.Int J Mol Sci. 2013 Jul 24;14(8):15423-58. doi: 10.3390/ijms140815423. Int J Mol Sci. 2013. PMID: 23887659 Free PMC article. Review.
-
The art of editing RNA structural alignments.Methods Mol Biol. 2014;1097:379-94. doi: 10.1007/978-1-62703-709-9_17. Methods Mol Biol. 2014. PMID: 24639168 Review.
Cited by
-
Robust and accurate prediction of noncoding RNAs from aligned sequences.BMC Bioinformatics. 2010 Oct 15;11 Suppl 7(Suppl 7):S3. doi: 10.1186/1471-2105-11-S7-S3. BMC Bioinformatics. 2010. PMID: 21106125 Free PMC article.
-
RNA Structure Elements Conserved between Mouse and 59 Other Vertebrates.Genes (Basel). 2018 Aug 1;9(8):392. doi: 10.3390/genes9080392. Genes (Basel). 2018. PMID: 30071678 Free PMC article.
-
Local conservation scores without a priori assumptions on neutral substitution rates.BMC Bioinformatics. 2008 Apr 11;9:190. doi: 10.1186/1471-2105-9-190. BMC Bioinformatics. 2008. PMID: 18405366 Free PMC article.
-
Uncovering deeply conserved motif combinations in rapidly evolving noncoding sequences.Genome Biol. 2021 Jan 11;22(1):29. doi: 10.1186/s13059-020-02247-1. Genome Biol. 2021. PMID: 33430943 Free PMC article.
-
Widespread purifying selection on RNA structure in mammals.Nucleic Acids Res. 2013 Sep;41(17):8220-36. doi: 10.1093/nar/gkt596. Epub 2013 Jul 11. Nucleic Acids Res. 2013. PMID: 23847102 Free PMC article.
References
-
- Margulies EH, Cooper GM, Asimenos G, Thomas DJ, Dewey CN, Siepel A, Birney E, Keefe D, Schwartz AS, Hou M, Taylor J, Nikolaev S, Montoya-Burgos JI, Löytynoja A, Whelan S, Pardi F, Massingham T, Brown JB, Bickel P, Holmes I, Mullikin JC, Ureta-Vidal A, Paten B, Stone EA, Rosenbloom KR, Kent WJ, Bouffard GG, Guan X, Hansen NF, Idol JR, Maduro VV, Maskeri B, McDowell JC, Park M, Thomas PJ, Young AC, Blakesley RW, Muzny DM, Sodergren E, Wheeler DA, Worley KC, Jiang H, Weinstock GM, Gibbs RA, Graves T, Fulton R, Mardis ER, Wilson RK, Clamp M, Cuff J, Gnerre S, Jaffie DB, Chang JL, Lindblad-Toh K, Lander ES, Hinrichs A, Trumbower H, Clawson H, Zweig A, Kuhn RM, Barber G, Harte R, Karolchik D, Field MA, Moore RA, Matthewson CA, Schein JE, Marra MA, Antonarakis SE, Batzoglou S, Goldman N, Hardison R, Haussler D, Miller W, Pachter L, Green ED, Sidow A. Analyses of deep mammalian sequence alignments and constraint predictions for 1% of the human genome. Genome Res. 2007;17:760–774. doi: 10.1101/gr.6034307. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
