

Astrocyte metabolism and signaling during brain ischemia
- PMID: 17965658
- PMCID: PMC8906499
- DOI: 10.1038/nn2004
Astrocyte metabolism and signaling during brain ischemia
Abstract
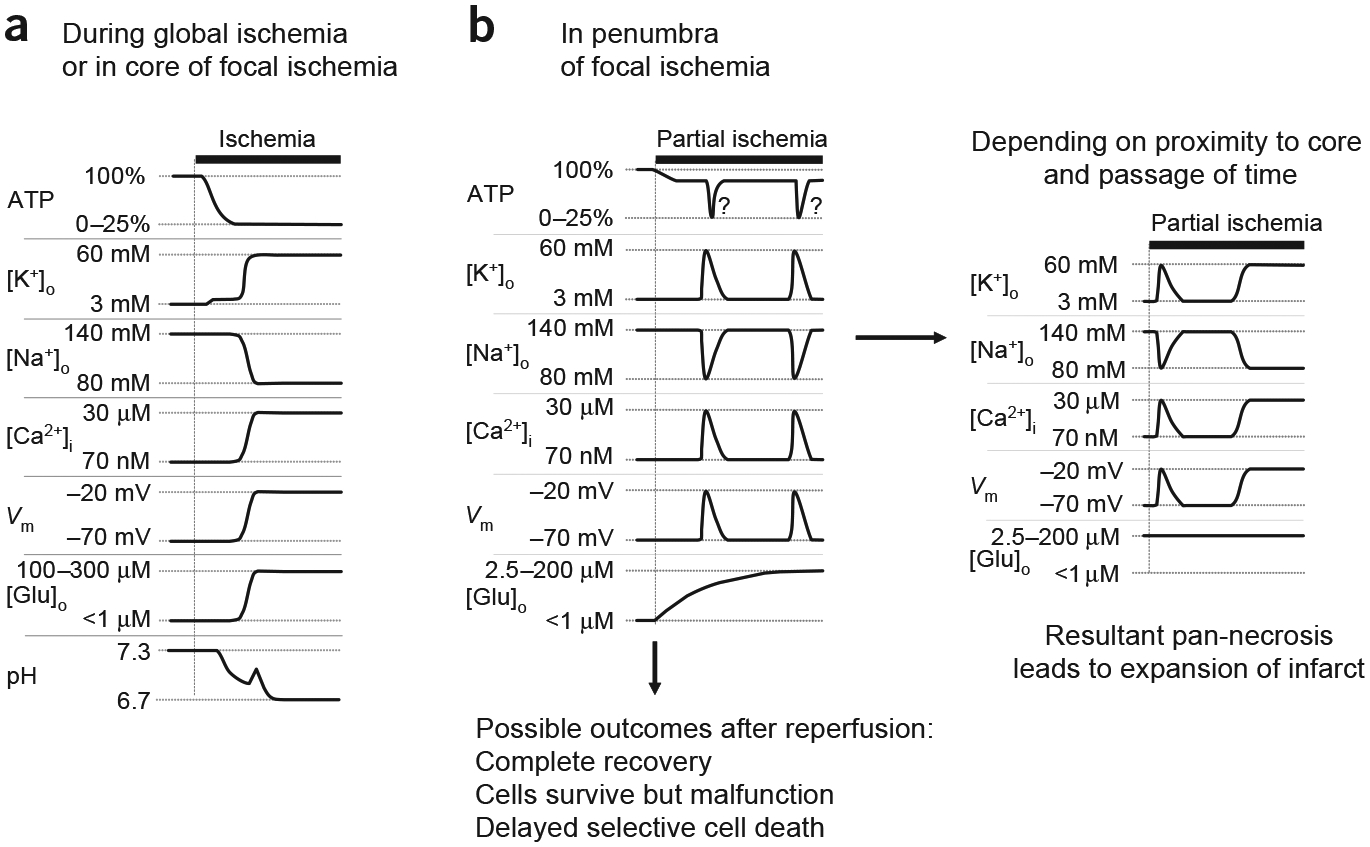
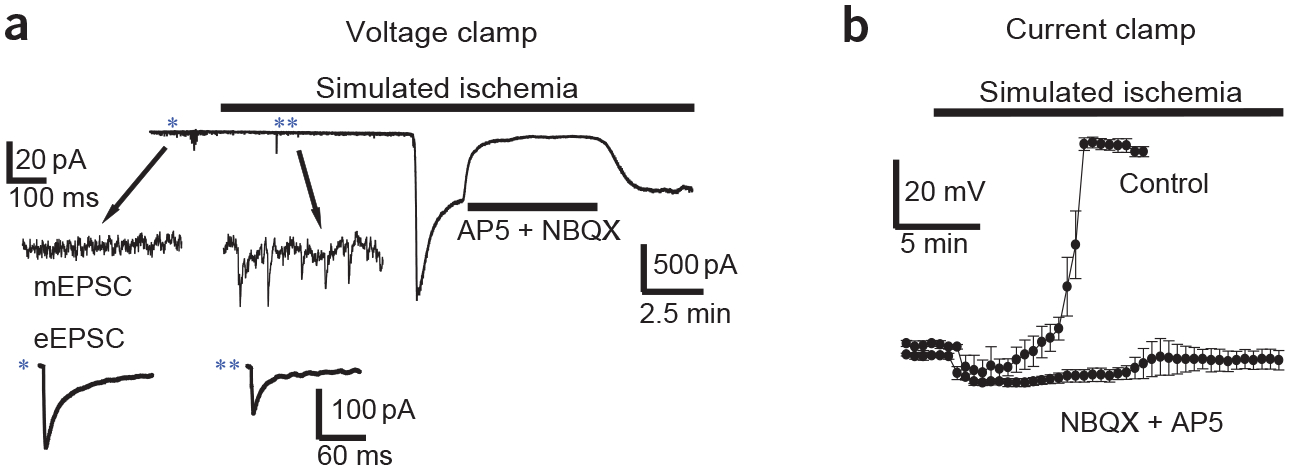
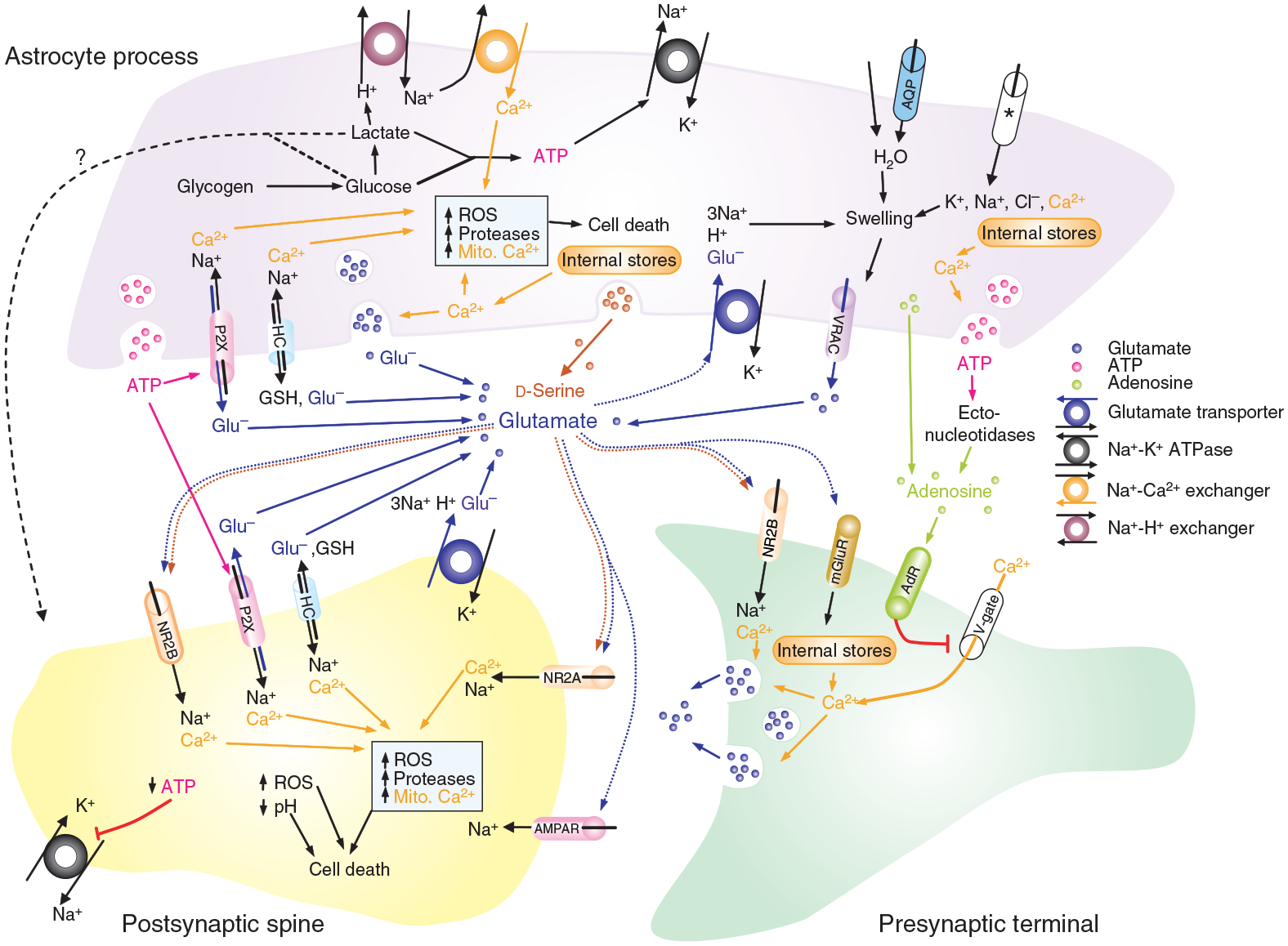
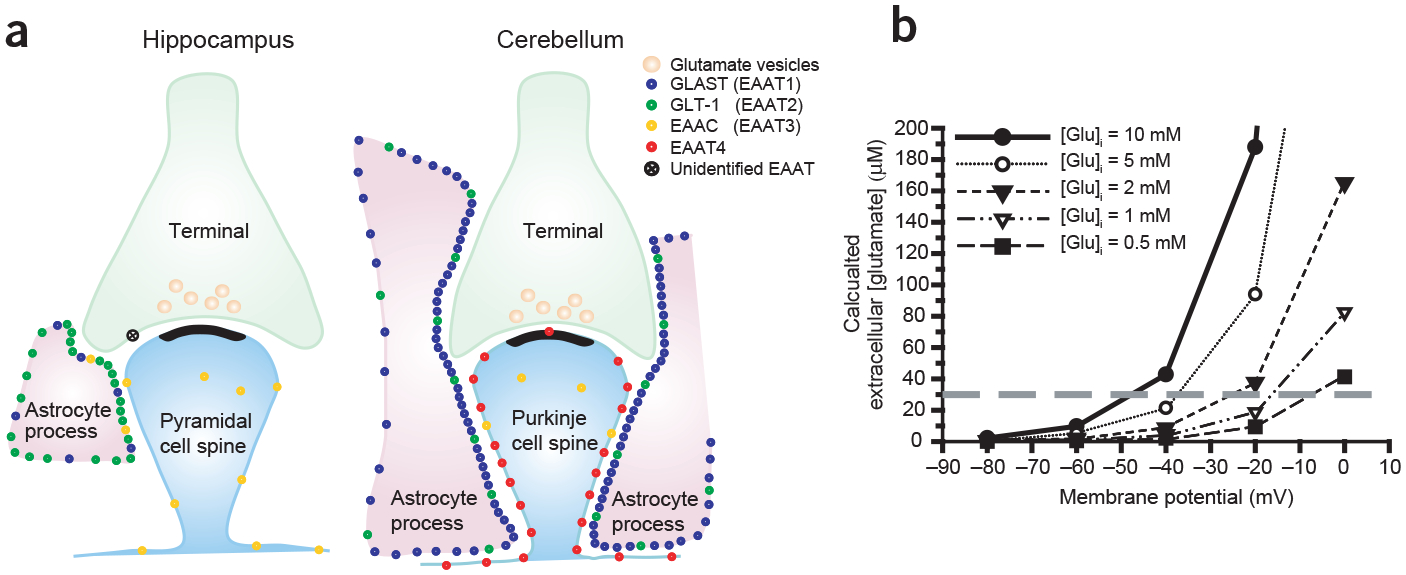
Brain ischemia results from cardiac arrest, stroke or head trauma. These conditions can cause severe brain damage and are a leading cause of death and long-term disability. Neurons are far more susceptible to ischemic damage than neighboring astrocytes, but astrocytes have diverse and important functions in many aspects of ischemic brain damage. Here we review three main roles of astrocytes in ischemic brain damage. First, we consider astrocyte glycogen stores, which can defend the brain against hypoglycemic brain damage but may aggravate brain damage during ischemia due to enhanced lactic acidosis. Second, we review recent breakthroughs in understanding astrocytic mechanisms of transmitter release, particularly for those transmitters with known roles in ischemic brain damage: glutamate, D-serine, ATP and adenosine. Third, we discuss the role of gap-junctionally connected networks of astrocytes in mediating the spread of damaging molecules to healthy 'bystanders' during infarct expansion in stroke.
Figures

References
-
- Choi DW & Rothman SM The role of glutamate neurotoxicity in hypoxic-ischemic neuronal death. Annu. Rev. Neurosci 13, 171–182 (1990). - PubMed
-
- Lipton P Ischemic cell death in brain neurons. Physiol. Rev 79, 1431–1568 (1999). - PubMed
-
- Silver IA, Deas J & Erecinska M Ion homeostasis in brain cells: differences in intracellular ion responses to energy limitation between cultured neurons and glial cells. Neuroscience 78, 589–601 (1997). - PubMed
-
- Lowry OH, Passonneau JV, Hasselberger FX & Schulz DW Effect of ischemia on known substrates and cofactors of the glycolytic pathway in brain. J. Biol. Chem 239, 18–30 (1964). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
