

Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome
- PMID: 17965736
- PMCID: PMC2241789
- DOI: 10.1038/sj.bjp.0707537
Coexistence of hERG current block and disruption of protein trafficking in ketoconazole-induced long QT syndrome
Abstract
Background and purpose: Many drugs associated with acquired long QT syndrome (LQTS) directly block human ether-a-go-go-related gene (hERG) K(+) channels. Recently, disrupted trafficking of the hERG channel protein was proposed as a new mechanism underlying LQTS, but whether this defect coexists with the hERG current block remains unclear. This study investigated how ketoconazole, a direct hERG current inhibitor, affects the trafficking of hERG channel protein.
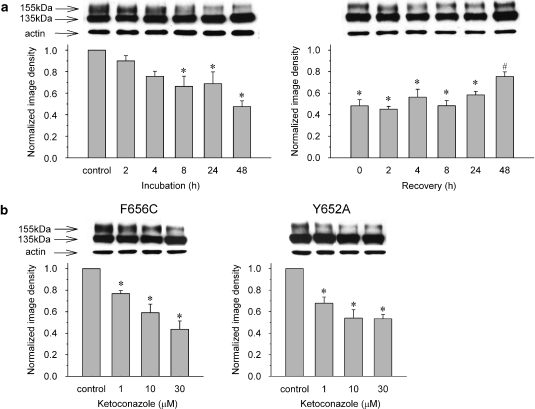
Experimental approach: Wild-type hERG and SCN5A/hNa(v) 1.5 Na(+) channels or the Y652A and F656C mutated forms of the hERG were stably expressed in HEK293 cells. The K(+) and Na(+) currents were recorded in these cells by using the whole-cell patch-clamp technique (23 degrees C). Protein trafficking of the hERG was evaluated by Western blot analysis and flow cytometry.
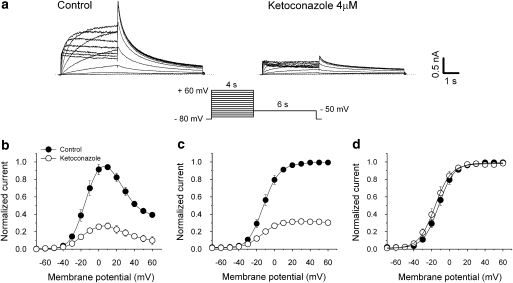
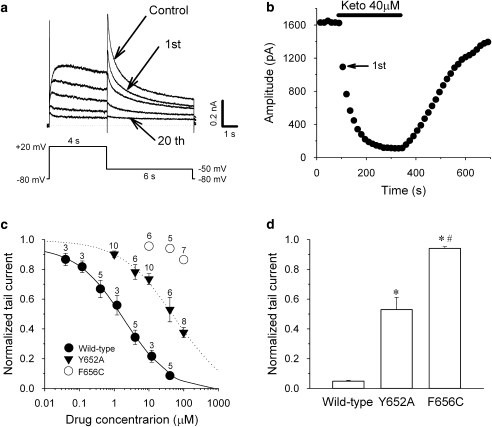
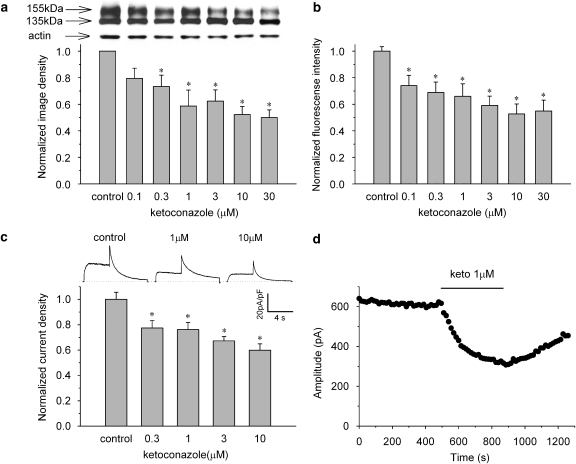
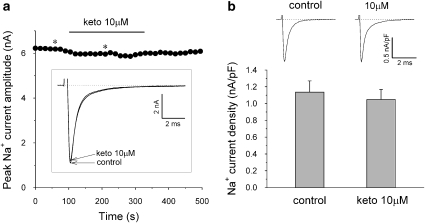
Key results: Ketoconazole directly blocked the hERG channel current and reduced the amount of hERG channel protein trafficked to the cell surface in a concentration-dependent manner. Current density of the hERG channels but not of the hNa(v) 1.5 channels was reduced after 48 h of incubation with ketoconazole, with preservation of the acute direct effect on hERG current. Mutations in drug-binding sites (F656C or Y652A) of the hERG channel significantly attenuated the hERG current blockade by ketoconazole, but did not affect the disruption of trafficking.
Conclusions and implications: Our findings indicate that ketoconazole might cause acquired LQTS via a direct inhibition of current through the hERG channel and by disrupting hERG protein trafficking within therapeutic concentrations. These findings should be considered when evaluating new drugs.
Figures
Comment in
-
Drugs and trafficking of ion channels: a new pro-arrhythmic threat on the horizon?Br J Pharmacol. 2008 Feb;153(3):406-9. doi: 10.1038/sj.bjp.0707618. Epub 2007 Dec 3. Br J Pharmacol. 2008. PMID: 18059314 Free PMC article.
References
-
- Albengres E, Le Louet H, Tillement JP. Systemic antifungal agents. Drug interactions of clinical significance. Drug Saf. 1998;18:83–97. - PubMed
-
- Carmeliet E. Mechanisms and control of repolarization. Eur Heart J. 1993;14 Suppl H:3–13. - PubMed
-
- Como JA, Dismukes WE. Oral azole drugs as systemic antifungal therapy. N Engl J Med. 1994;330:263–272. - PubMed
-
- Curran ME, Splawski I, Timothy KW, Vincent GM, Green ED, Keating MT. A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell. 1995;80:795–803. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Miscellaneous
