

Antagonism of thromboxane receptors by diclofenac and lumiracoxib
- PMID: 17965743
- PMCID: PMC2189986
- DOI: 10.1038/sj.bjp.0707518
Antagonism of thromboxane receptors by diclofenac and lumiracoxib
Erratum in
- Br J Pharmacol. 2008 Apr;153(8):1763. Ambrosio, E [corrected to Ambrosio, M]
Abstract
Background and purpose: Non-steroidal anti-inflammatory drugs (NSAIDs) are analgesic and anti-inflammatory by virtue of inhibition of the cyclooxygenase (COX) reaction that initiates biosynthesis of prostaglandins. Findings in a pulmonary pharmacology project gave rise to the hypothesis that certain members of the NSAID class might also be antagonists of the thromboxane (TP) receptor.
Experimental approach: Functional responses due to activation of the TP receptor were studied in isolated airway and vascular smooth muscle preparations from guinea pigs and rats as well as in human platelets. Receptor binding and activation of the TP receptor was studied in HEK293 cells.
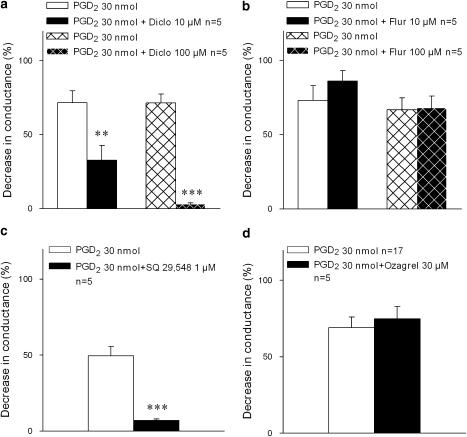
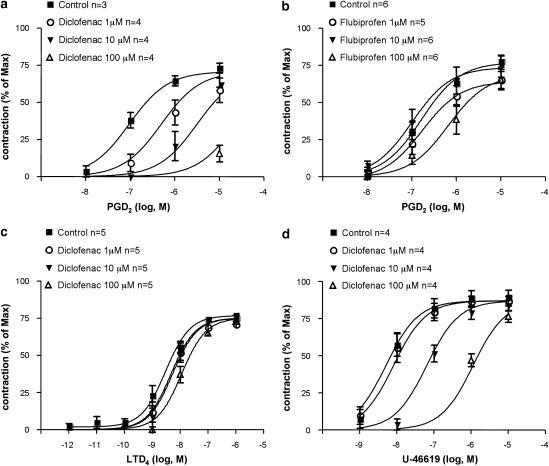
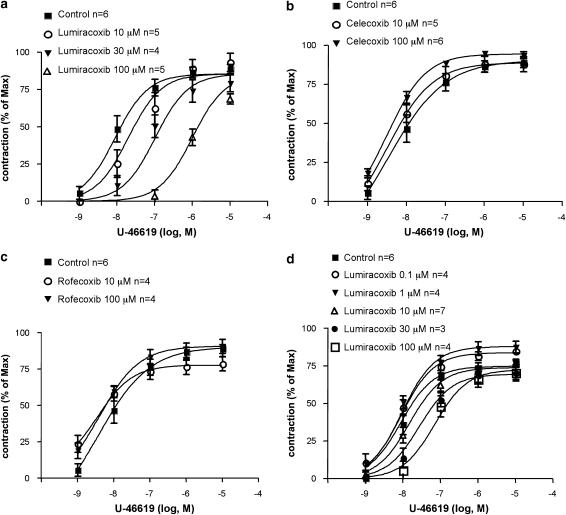
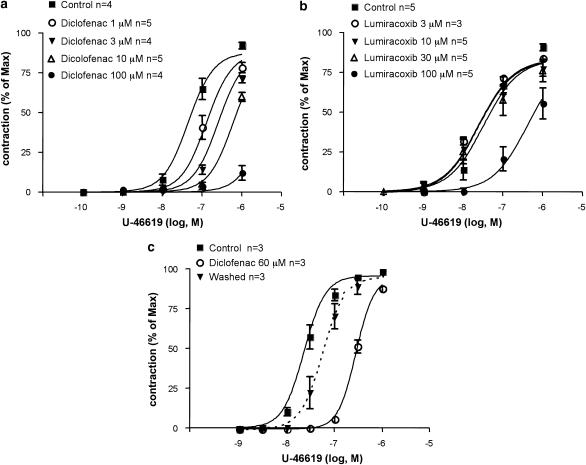
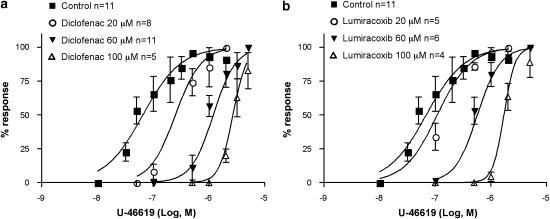
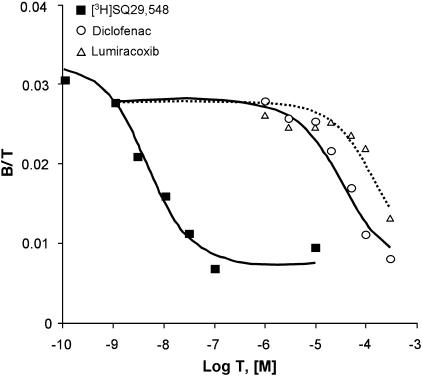
Key results: Diclofenac concentration-dependently and selectively inhibited the contraction responses to TP receptor agonists such as prostaglandin D2 and U-46619 in the tested smooth muscle preparations and the aggregation of human platelets. The competitive antagonism of the TP receptor was confirmed by binding studies and at the level of signal transduction. The selective COX-2 inhibitor lumiracoxib shared this activity profile, whereas a number of standard NSAIDs and other selective COX-2 inhibitors did not.
Conclusions and implications: Diclofenac and lumiracoxib, in addition to being COX unselective and highly COX-2 selective inhibitors, respectively, displayed a previously unknown pharmacological activity, namely TP receptor antagonism. Development of COX-2 selective inhibitors with dual activity as potent TP antagonists may lead to coxibs with improved cardiovascular safety, as the TP receptor mediates cardiovascular effects of thromboxane A2 and isoprostanes.
Figures

References
-
- Audoly LP, Rocca B, Fabre JE, Koller BH, Thomas D, Loeb AL, et al. Cardiovascular responses to the isoprostanes iPF(2alpha)-III and iPE(2)-III are mediated via the thromboxane A(2) receptor in vivo. Circulation. 2000;101:2833–2840. - PubMed
-
- Bombardier C, Laine L, Reicin A, Shapiro D, Burgos-Vargas R, Davis B, et al. Comparison of upper gastrointestinal toxicity of rofecoxib and naproxen in patients with rheumatoid arthritis. VIGOR Study Group. N Engl J Med. 2000;343:1520–1528. - PubMed
-
- Bresalier RS, Sandler RS, Quan H, Bolognese JA, Oxenius B, Horgan K, et al. Cardiovascular events associated with rofecoxib in a colorectal adenoma chemoprevention trial. N Engl J Med. 2005;352:1092–1102. - PubMed
-
- Capra V, Habib A, Accomazzo MR, Ravasi S, Citro S, Levy-Toledano S, et al. Thromboxane prostanoid receptor in human airway smooth muscle cells: a relevant role in proliferation. Eur J Pharmacol. 2003;474:149–159. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
