

Construction, visualisation, and clustering of transcription networks from microarray expression data
- PMID: 17967053
- PMCID: PMC2041979
- DOI: 10.1371/journal.pcbi.0030206
Construction, visualisation, and clustering of transcription networks from microarray expression data
Abstract
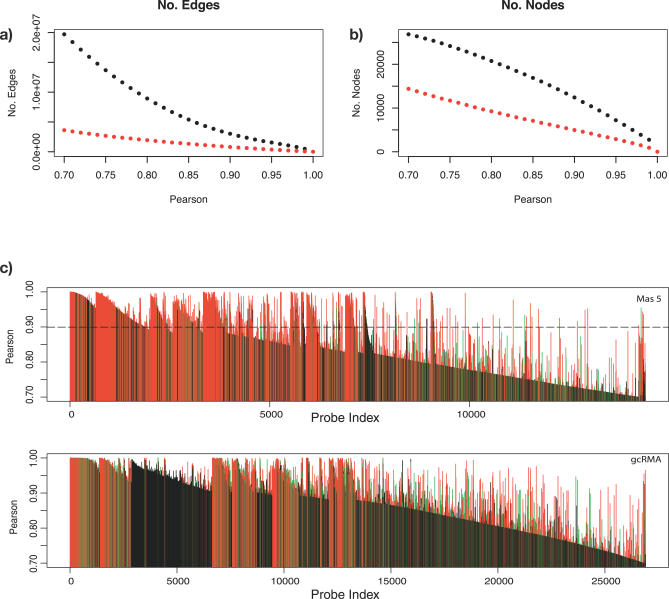
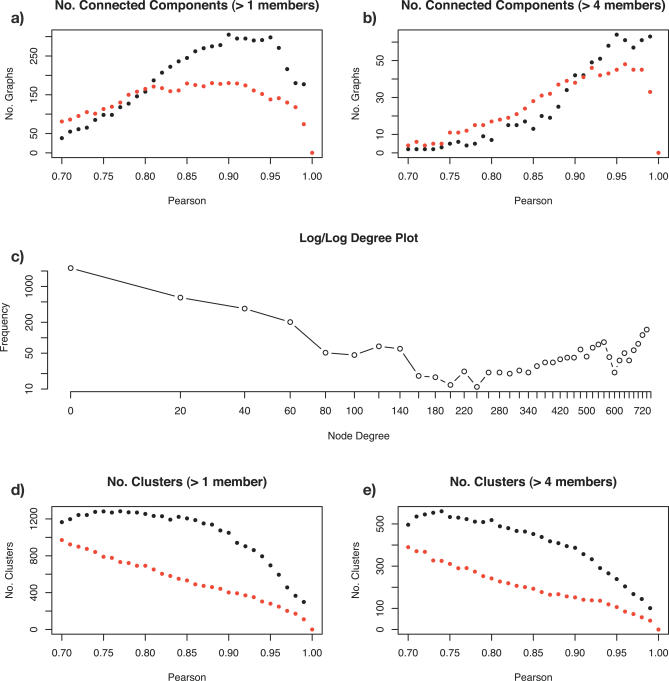
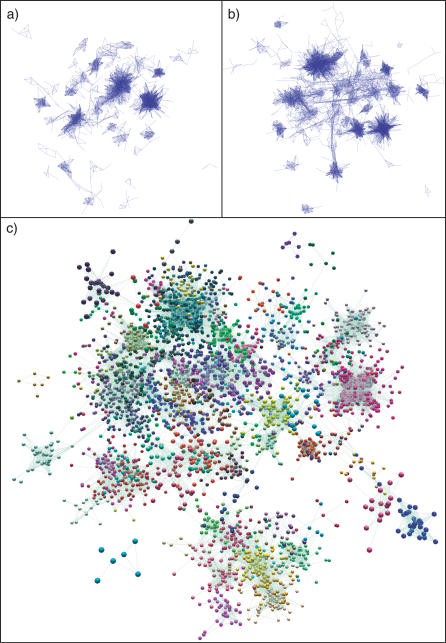
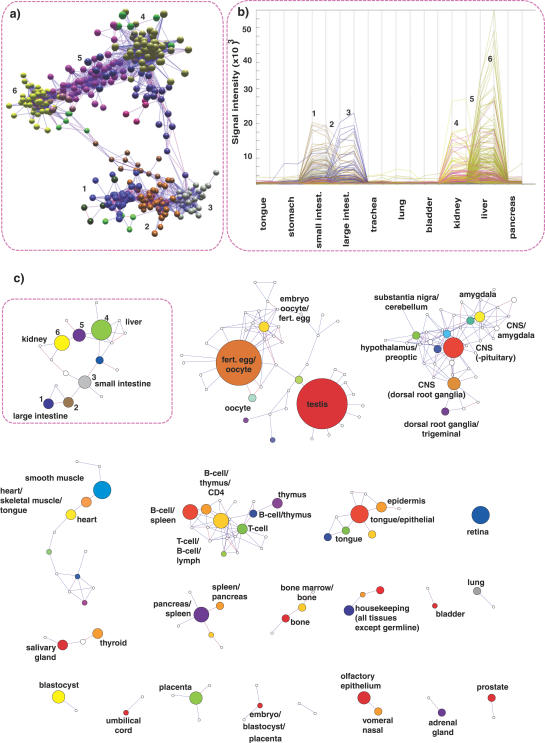
Network analysis transcends conventional pairwise approaches to data analysis as the context of components in a network graph can be taken into account. Such approaches are increasingly being applied to genomics data, where functional linkages are used to connect genes or proteins. However, while microarray gene expression datasets are now abundant and of high quality, few approaches have been developed for analysis of such data in a network context. We present a novel approach for 3-D visualisation and analysis of transcriptional networks generated from microarray data. These networks consist of nodes representing transcripts connected by virtue of their expression profile similarity across multiple conditions. Analysing genome-wide gene transcription across 61 mouse tissues, we describe the unusual topography of the large and highly structured networks produced, and demonstrate how they can be used to visualise, cluster, and mine large datasets. This approach is fast, intuitive, and versatile, and allows the identification of biological relationships that may be missed by conventional analysis techniques. This work has been implemented in a freely available open-source application named BioLayout Express(3D).
Conflict of interest statement
Figures
References
-
- Reed JL, Famili I, Thiele I, Palsson BO. Towards multidimensional genome annotation. Nat Rev Genet. 2006;7:130–141. - PubMed
-
- Kitano H. Computational systems biology. Nature. 2002;420:206–210. - PubMed
-
- Nurse P. Systems biology: Understanding cells. Nature. 2003;424:883. - PubMed
-
- Cassman M. Barriers to progress in systems biology. Nature. 2005;438:1079. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
