

Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters
- PMID: 17967063
- PMCID: PMC2041996
- DOI: 10.1371/journal.pgen.0030181
Genome-wide profiling of DNA methylation reveals a class of normally methylated CpG island promoters
Abstract
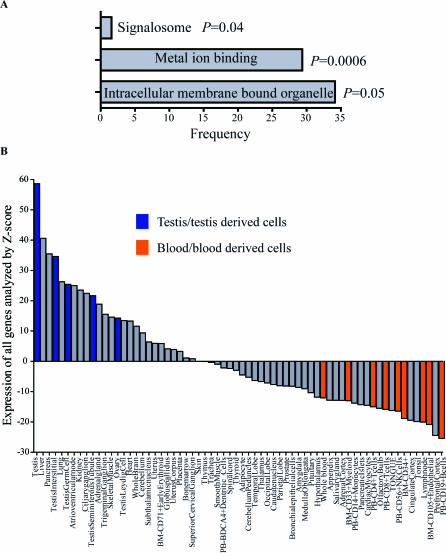
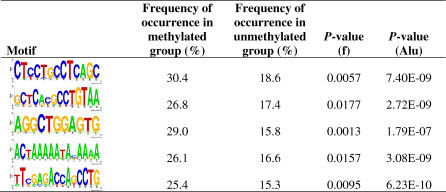
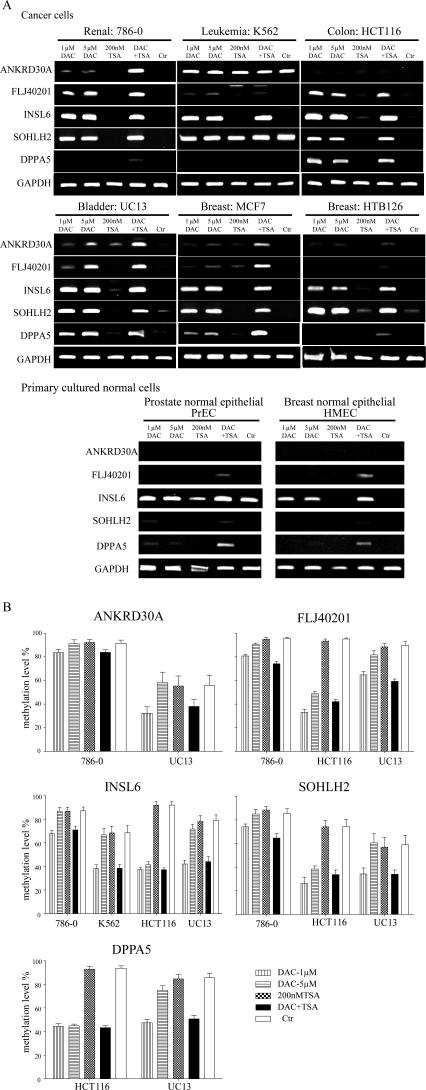
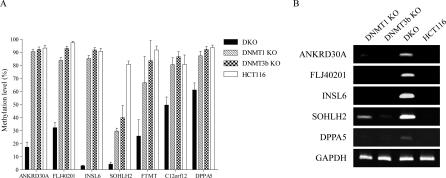
The role of CpG island methylation in normal development and cell differentiation is of keen interest, but remains poorly understood. We performed comprehensive DNA methylation profiling of promoter regions in normal peripheral blood by methylated CpG island amplification in combination with microarrays. This technique allowed us to simultaneously determine the methylation status of 6,177 genes, 92% of which include dense CpG islands. Among these 5,549 autosomal genes with dense CpG island promoters, we have identified 4.0% genes that are nearly completely methylated in normal blood, providing another exception to the general rule that CpG island methylation in normal tissue is limited to X inactivation and imprinted genes. We examined seven genes in detail, including ANKRD30A, FLJ40201, INSL6, SOHLH2, FTMT, C12orf12, and DPPA5. Dense promoter CpG island methylation and gene silencing were found in normal tissues studied except testis and sperm. In both tissues, bisulfite cloning and sequencing identified cells carrying unmethylated alleles. Interestingly, hypomethylation of several genes was associated with gene activation in cancer. Furthermore, reactivation of silenced genes could be induced after treatment with a DNA demethylating agent or in a cell line lacking DNMT1 and/or DNMT3b. Sequence analysis identified five motifs significantly enriched in this class of genes, suggesting that cis-regulatory elements may facilitate preferential methylation at these promoter CpG islands. We have identified a group of non-X-linked bona fide promoter CpG islands that are densely methylated in normal somatic tissues, escape methylation in germline cells, and for which DNA methylation is a primary mechanism of tissue-specific gene silencing.
Conflict of interest statement
Competing interests. The authors have declared that no competing interests exist.
Figures




References
-
- Bird AP. Cpg-rich islands and the function of DNA methylation. Nature. 1986;321:209–213. - PubMed
-
- Razin A, Cedar H. DNA methylation and genomic imprinting. Cell. 1994;77:473–476. - PubMed
-
- Jaenisch R, Bird A. Epigenetic regulation of gene expression: how the genome integrates intrinsic and environmental signals. Nat Genet. 2003;33:245–254. - PubMed
-
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet. 2002;3:662–673. - PubMed
-
- Jones PA, Takai D. The role of DNA methylation in mammalian epigenetics. Science. 2001;293:1068–1070. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
