

A molecular switch that controls cell spreading and retraction
- PMID: 17967945
- PMCID: PMC2064799
- DOI: 10.1083/jcb.200703185
A molecular switch that controls cell spreading and retraction
Abstract
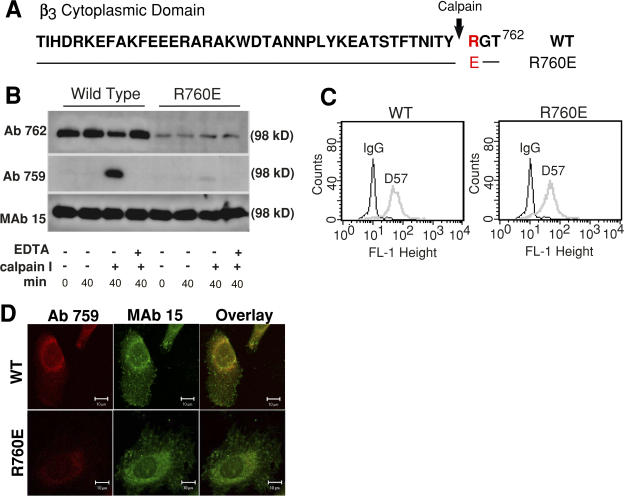
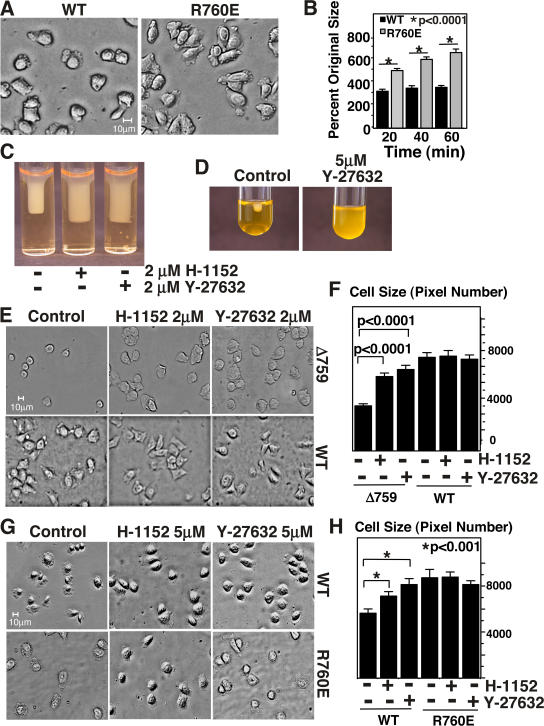
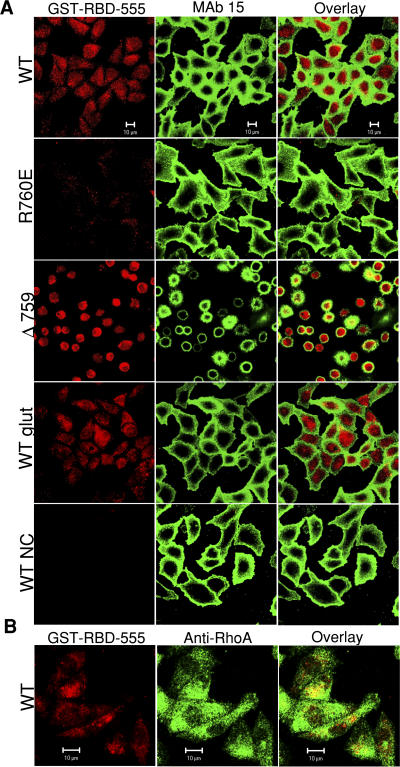
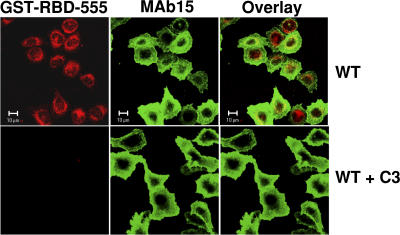
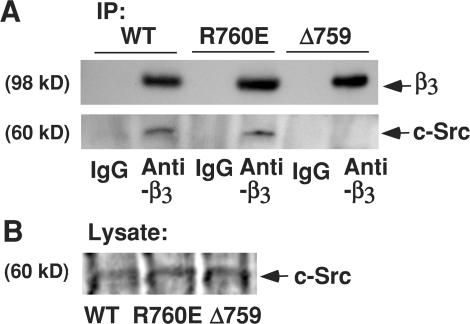
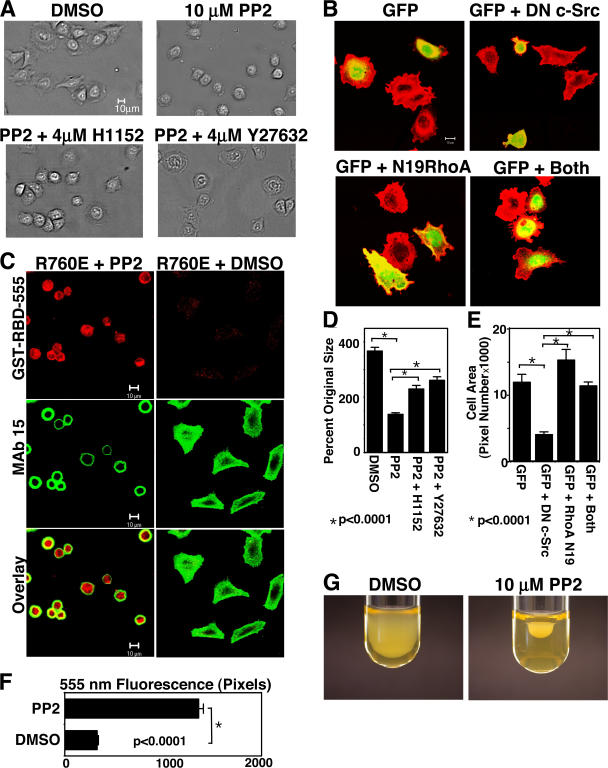
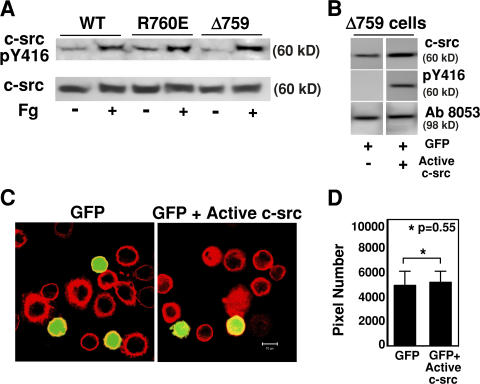
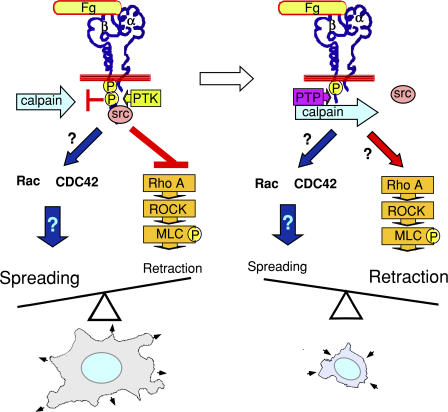
Integrin-dependent cell spreading and retraction are required for cell adhesion, migration, and proliferation, and thus are important in thrombosis, wound repair, immunity, and cancer development. It remains unknown how integrin outside-in signaling induces and controls these two opposite processes. This study reveals that calpain cleavage of integrin beta(3) at Tyr(759) switches the functional outcome of integrin signaling from cell spreading to retraction. Expression of a calpain cleavage-resistant beta(3) mutant in Chinese hamster ovary cells causes defective clot retraction and RhoA-mediated retraction signaling but enhances cell spreading. Conversely, a calpain-cleaved form of beta(3) fails to mediate cell spreading, but inhibition of the RhoA signaling pathway corrects this defect. Importantly, the calpain-cleaved beta(3) fails to bind c-Src, which is required for integrin-induced cell spreading, and this requirement of beta(3)-associated c-Src results from its inhibition of RhoA-dependent contractile signals. Thus, calpain cleavage of beta(3) at Tyr(759) relieves c-Src-mediated RhoA inhibition, activating the RhoA pathway that confines cell spreading and causes cell retraction.
Figures

References
-
- Amano, M., M. Ito, K. Kimura, Y. Fukata, K. Chihara, T. Nakano, Y. Matsuura, and K. Kaibuchi. 1996. Phosphorylation and activation of myosin by Rho-associated kinase (Rho-kinase). J. Biol. Chem. 271:20246–20249. - PubMed
-
- Arias-Salgado, E.G., S. Lizano, S.J. Shattil, and M.H. Ginsberg. 2005. b. Specification of the direction of adhesive signaling by the integrin beta cytoplasmic domain. J. Biol. Chem. 280:29699–29707. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
