

Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias
- PMID: 17967977
- PMCID: PMC3150966
- DOI: 10.1161/CIRCULATIONAHA.107.703330
Mutation in glycerol-3-phosphate dehydrogenase 1 like gene (GPD1-L) decreases cardiac Na+ current and causes inherited arrhythmias
Abstract
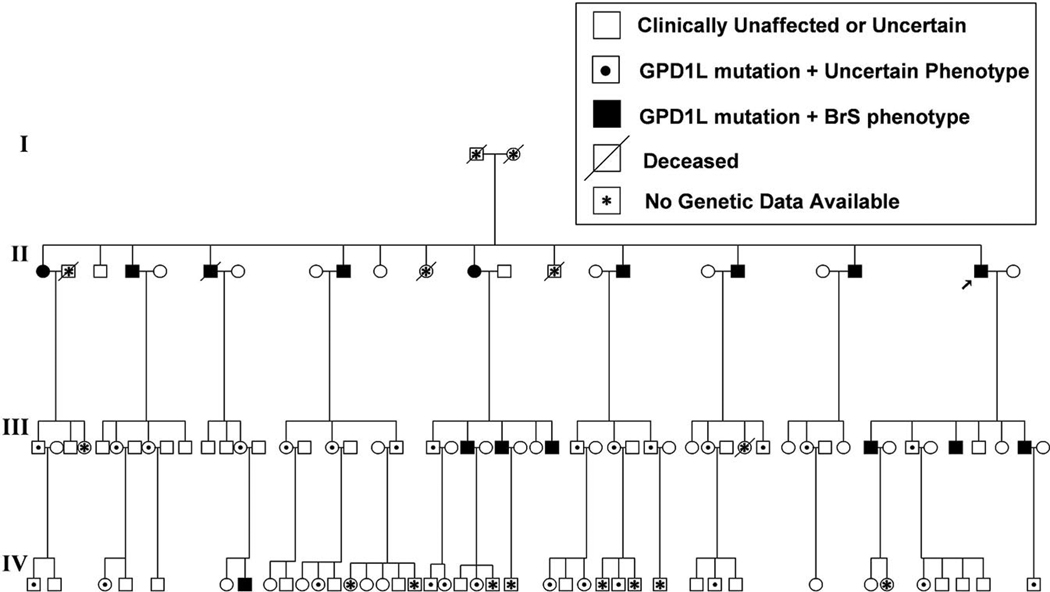
Background: Brugada syndrome is a rare, autosomal-dominant, male-predominant form of idiopathic ventricular fibrillation characterized by a right bundle-branch block and ST elevation in the right precordial leads of the surface ECG. Mutations in the cardiac Na+ channel SCN5A on chromosome 3p21 cause approximately 20% of the cases of Brugada syndrome; most mutations decrease inward Na+ current, some by preventing trafficking of the channels to the surface membrane. We previously used positional cloning to identify a new locus on chromosome 3p24 in a large family with Brugada syndrome and excluded SCN5A as a candidate gene.
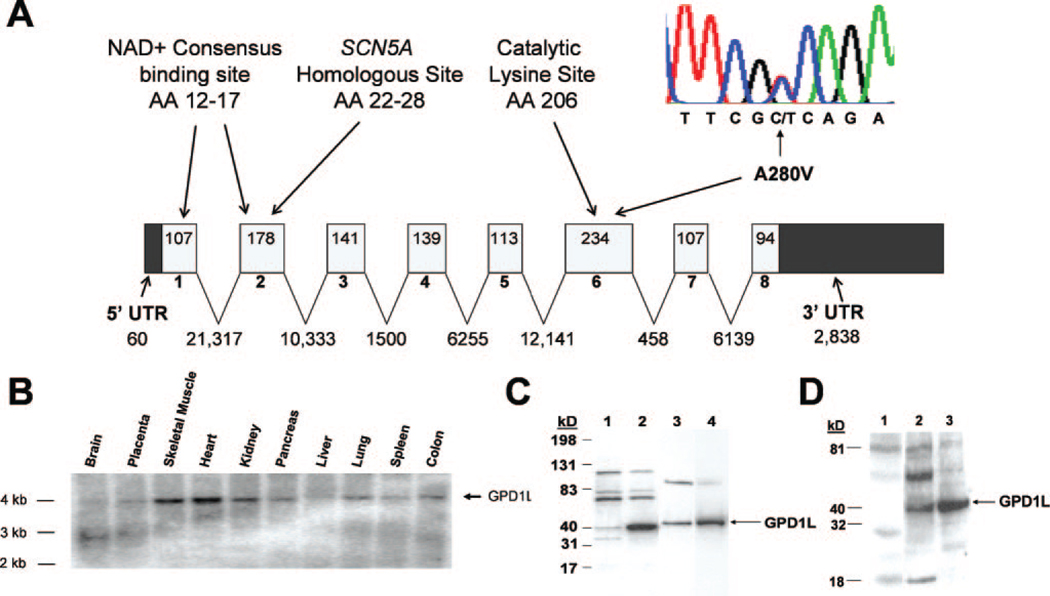
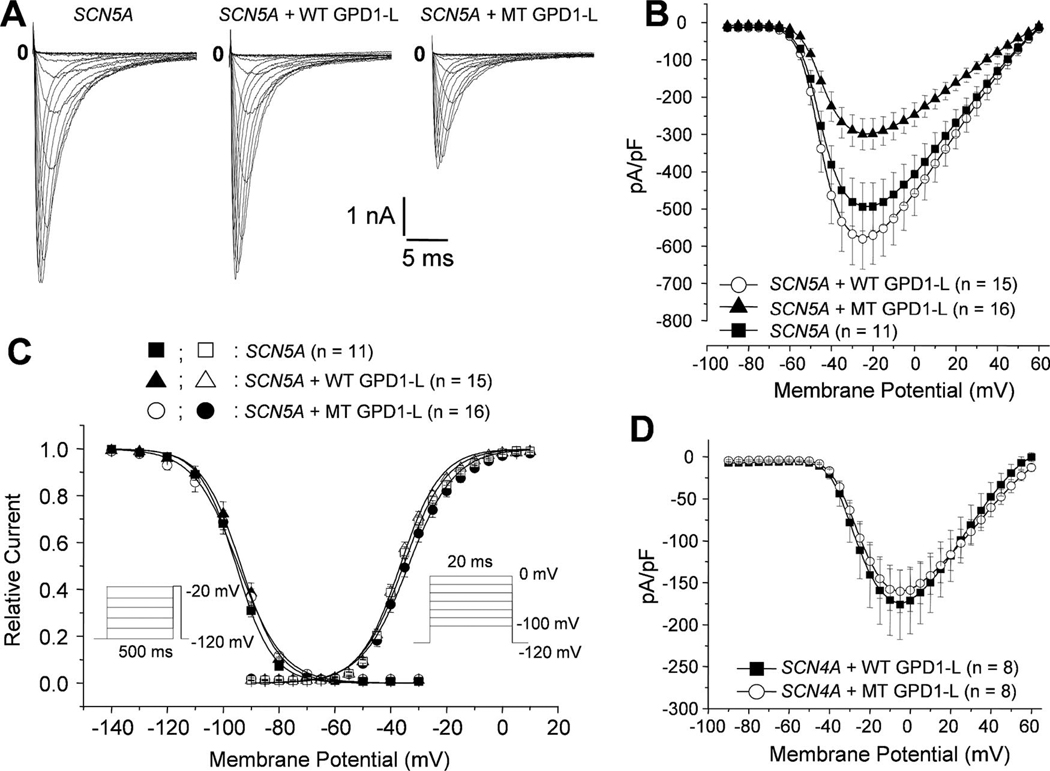
Methods and results: We used direct sequencing to identify a mutation (A280V) in a conserved amino acid of the glycerol-3-phosphate dehydrogenase 1-like (GPD1-L) gene. The mutation was present in all affected individuals and absent in >500 control subjects. GPD1-L RNA and protein are abundant in the heart. Compared with wild-type GPD1-L, coexpression of A280V GPD1-L with SCN5A in HEK cells reduced inward Na+ currents by approximately 50% (P<0.005). Wild-type GPD1-L localized near the cell surface to a greater extent than A280V GPD1-L. Coexpression of A280V GPD1-L with SCN5A reduced SCN5A cell surface expression by 31+/-5% (P=0.01).
Conclusions: GPD1-L is a novel gene that may affect trafficking of the cardiac Na+ channel to the cell surface. A GPD1-L mutation decreases SCN5A surface membrane expression, reduces inward Na+ current, and causes Brugada syndrome.
Figures


References
-
- Antzelevitch C, Brugada P, Borggrefe M, Brugada J, Brugada R, Corrado D, Gussak I, LeMarec H, Nademanee K, Perez Riera AR, Shimizu W, Schulze-Bahr E, Tan H, Wilde A. Brugada syndrome: report of the Second Consensus Conference: endorsed by the Heart Rhythm Society and the European Heart Rhythm Association. Circulation. 2005;111:659–670. - PubMed
-
- Brugada J, Brugada P. Further characterization of the syndrome of right bundle branch block, ST segment elevation, and sudden cardiac death. J Cardiovasc Electrophysiol. 1997;8:325–331. - PubMed
-
- Brugada P, Brugada J. Right bundle branch block, persistent ST segment elevation and sudden cardiac death: a distinct clinical and electrocardiographic syndrome: a multicenter report. J Am Coll Cardiol. 1992;20:1391–1396. - PubMed
-
- Grant AO. Electrophysiological basis and genetics of Brugada syndrome. J Cardiovasc Electrophysiol. 2005;16:S3–S7. - PubMed
-
- Chen Q, Kirsch GE, Zhang D, Brugada R, Brugada J, Brugada P, Potenza D, Moya A, Borggrefe M, Breithardt G, Ortiz-Lopez R, Wang Z, Antzelevitch C, O’Brien RE, Schulze-Bahr E, Keating MT, Towbin JA, Wang Q. Genetic basis and molecular mechanism for idiopathic ventricular fibrillation. Nature. 1998;392:293–296. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
