

Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium
- PMID: 17970581
- PMCID: PMC2602826
- DOI: 10.1021/tx700198a
Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium
Abstract
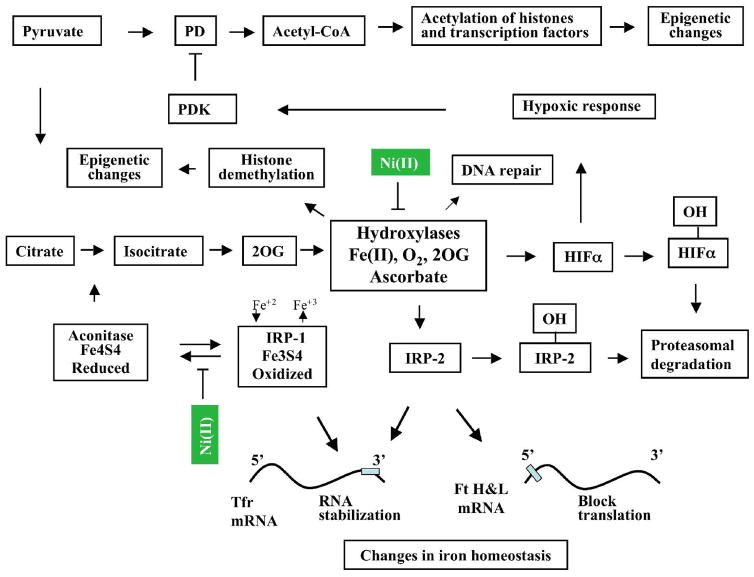
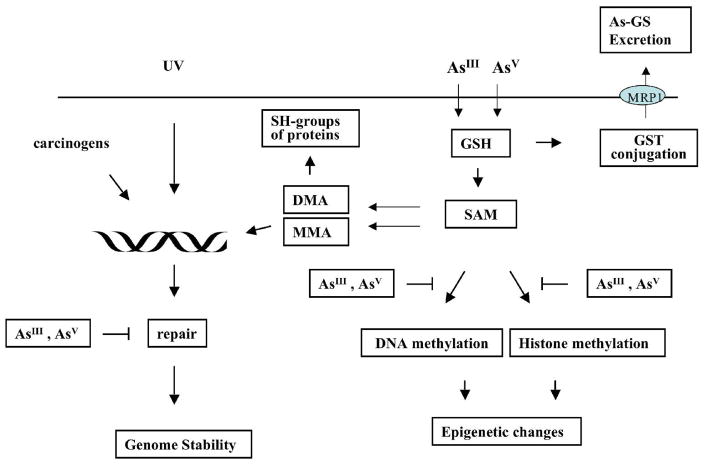
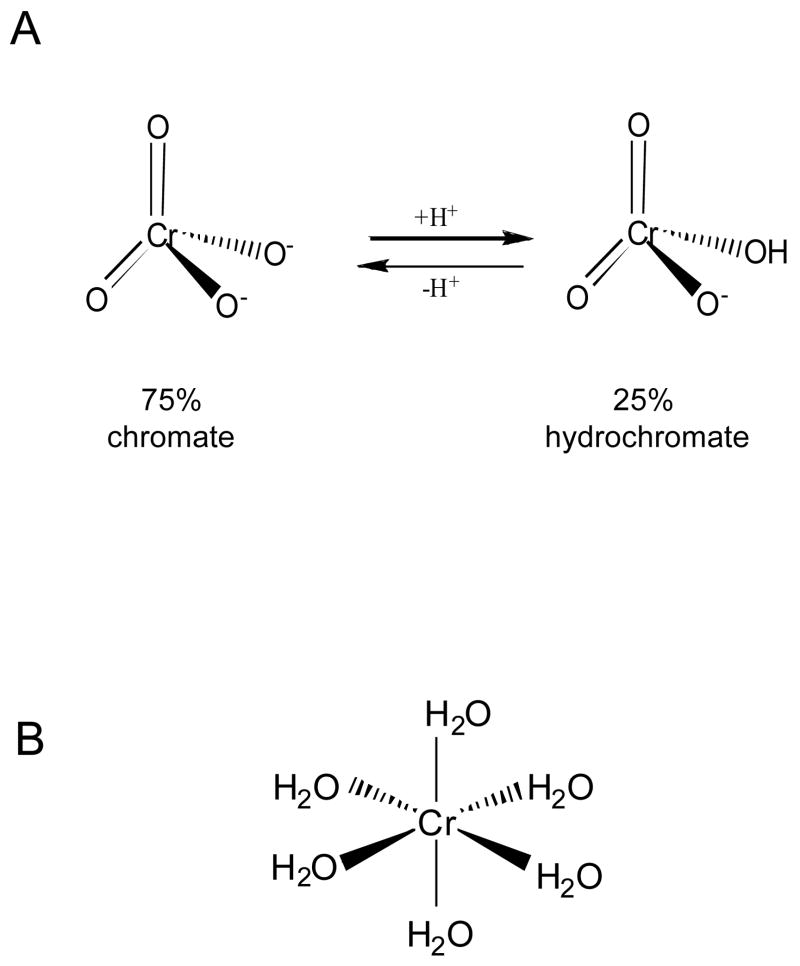
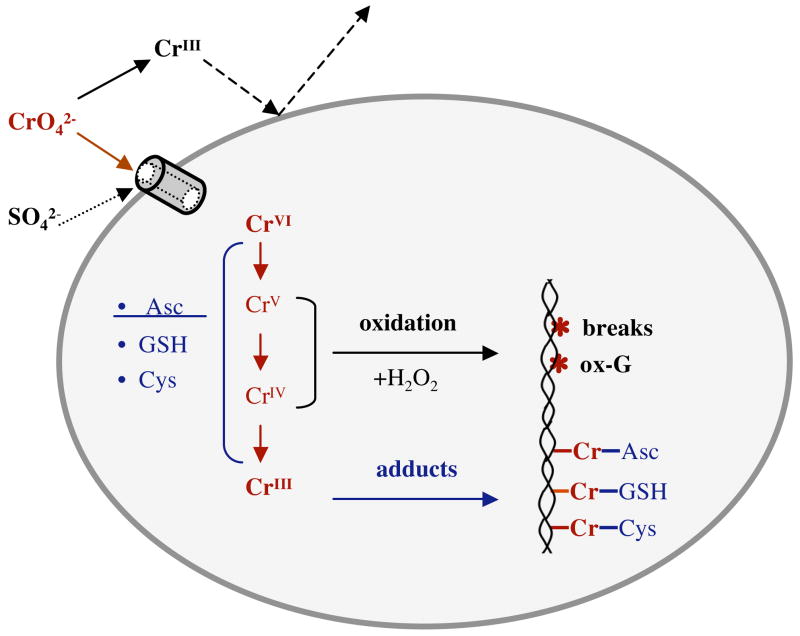
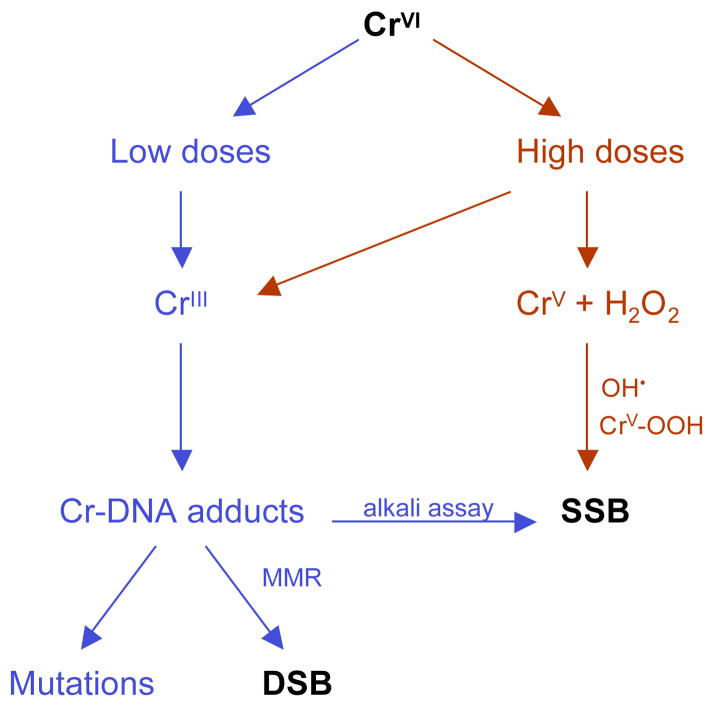
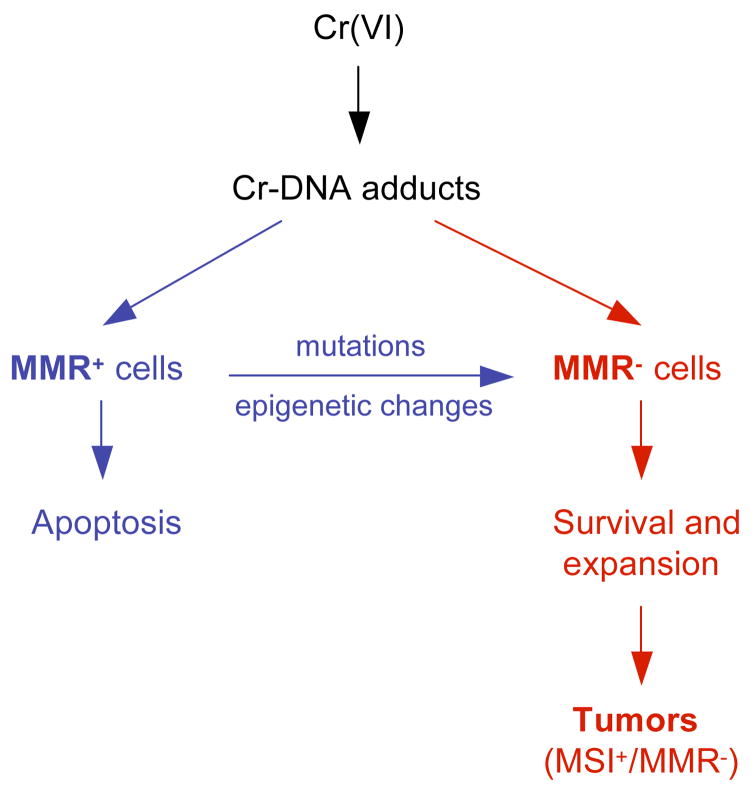
Chronic exposure to nickel(II), chromium(VI), or inorganic arsenic (iAs) has long been known to increase cancer incidence among affected individuals. Recent epidemiological studies have found that carcinogenic risks associated with chromate and iAs exposures were substantially higher than previously thought, which led to major revisions of the federal standards regulating ambient and drinking water levels. Genotoxic effects of Cr(VI) and iAs are strongly influenced by their intracellular metabolism, which creates several reactive intermediates and byproducts. Toxic metals are capable of potent and surprisingly selective activation of stress-signaling pathways, which are known to contribute to the development of human cancers. Depending on the metal, ascorbate (vitamin C) has been found to act either as a strong enhancer or suppressor of toxic responses in human cells. In addition to genetic damage via both oxidative and nonoxidative (DNA adducts) mechanisms, metals can also cause significant changes in DNA methylation and histone modifications, leading to epigenetic silencing or reactivation of gene expression. In vitro genotoxicity experiments and recent animal carcinogenicity studies provided strong support for the idea that metals can act as cocarcinogens in combination with nonmetal carcinogens. Cocarcinogenic and comutagenic effects of metals are likely to stem from their ability to interfere with DNA repair processes. Overall, metal carcinogenesis appears to require the formation of specific metal complexes, chromosomal damage, and activation of signal transduction pathways promoting survival and expansion of genetically/epigenetically altered cells.
Figures
References
-
- Kasprzak KS, Sunderman FW, Salnikow K. Nickel carcinogenesis. Mutat Res. 2003;533:67–97. - PubMed
-
- Morgan LG, Usher V. Health problems associated with nickel refining and use. Ann Occup Hyg. 1994;38:189–198. - PubMed
-
- Sunderman FW., Jr Carcinogenicity of nickel compounds in animals. IARC Sci Publ. 1984:127–142. - PubMed
-
- Sutherland JE, Costa M. Epigenetics and the environment. Ann N Y Acad Sci. 2003;983:151–160. - PubMed
-
- Lu H, Shi X, Costa M, Huang C. Carcinogenic effect of nickel compounds. Mol Cell Biochem. 2005;279:45–67. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
