

Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway
- PMID: 17971904
- PMCID: PMC2040211
- DOI: 10.1593/neo.07517
Molecular analysis of a multistep lung cancer model induced by chronic inflammation reveals epigenetic regulation of p16 and activation of the DNA damage response pathway
Abstract
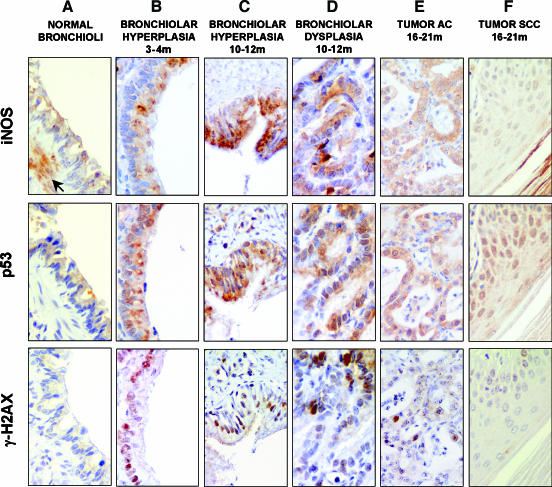
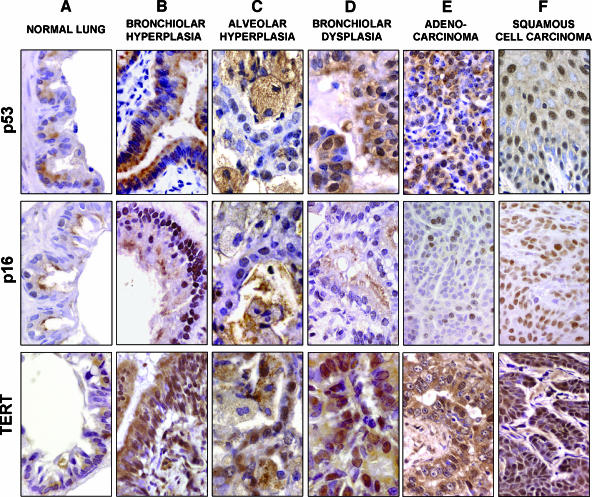
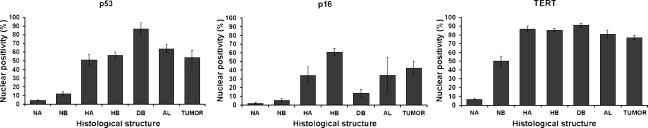
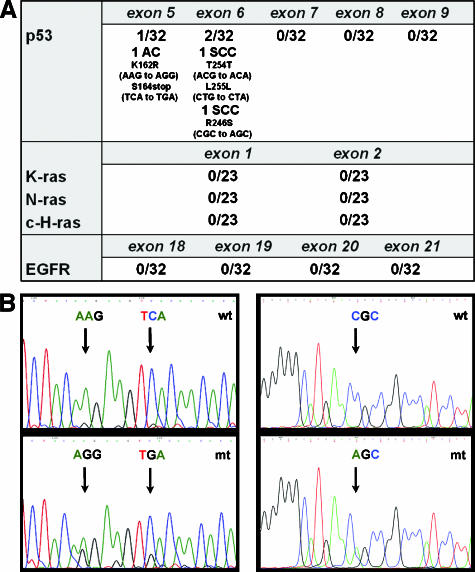
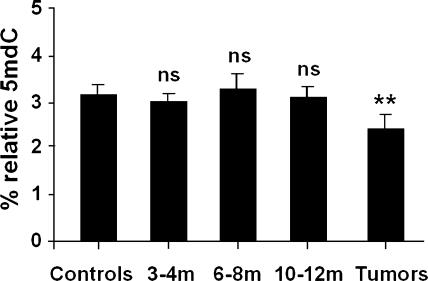
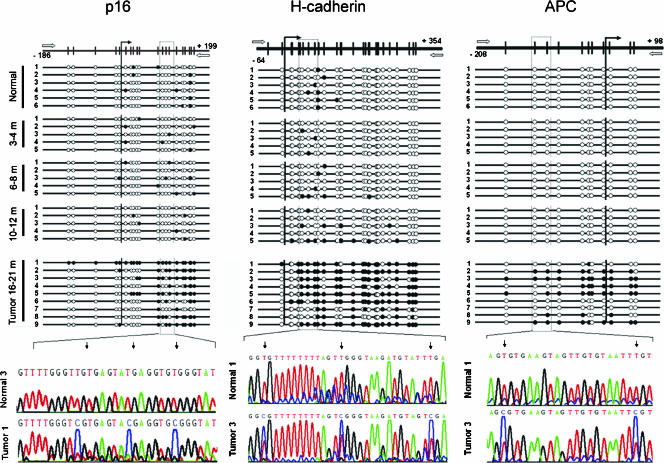
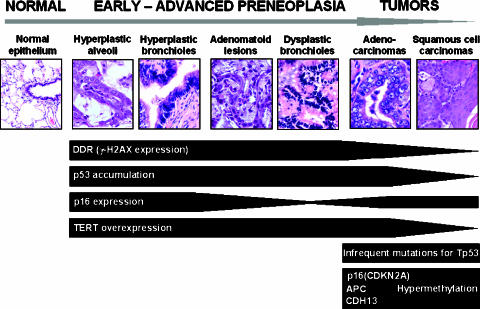
The molecular hallmarks of inflammation-mediated lung carcinogenesis have not been fully clarified, mainly due to the scarcity of appropriate animal models. We have used a silica-induced multistep lung carcinogenesis model driven by chronic inflammation to study the evolution of molecular markers and genetic alterations. We analyzed markers of DNA damage response (DDR), proliferative stress, and telomeric stress: gamma-H2AX, p16, p53, and TERT. Lung cancer-related epigenetic and genetic alterations, including promoter hypermethylation status of p16(CDKN2A), APC, CDH13, Rassf1, and Nore1A, as well as mutations of Tp53, epidermal growth factor receptor, K-ras, N-ras, and c-H-ras, have been also studied. Our results showed DDR pathway activation in preneoplastic lesions, in association with inducible nitric oxide synthase and p53 induction. p16 was also induced in early tumorigenic progression and was inactivated in bronchiolar dysplasias and tumors. Remarkably, lack of mutations of Ras and epidermal growth factor receptor, and a very low frequency of Tp53 mutations suggest that they are not required for tumorigenesis in this model. In contrast, epigenetic alterations in p16(CDKN2A), CDH13, and APC, but not in Rassf1 and Nore1A, were clearly observed. These data suggest the existence of a specific molecular signature of inflammation-driven lung carcinogenesis that shares some, but not all, of the molecular landmarks of chemically induced lung cancer.
Keywords: DNA damage response; animal model; inflammation; lung cancer; preneoplastic lesions.
Figures
References
-
- Sato M, Shames DS, Gazdar AF, Minna JD. A translational view of the molecular pathogenesis of lung cancer. J Thorac Oncol. 2007;2:327–343. - PubMed
-
- Hecht SS. Tobacco carcinogens, their biomarkers and tobacco-induced cancer. Nat Rev Cancer. 2003;3:733–744. - PubMed
-
- Hammond SK, Sorensen G, Youngstrom R, Ockene JK. Occupational exposure to environmental tobacco smoke. JAMA. 1995;274:956–960. - PubMed
-
- Dalgleish A, Haefner B, editors. Wounds That Do Not Heal. New York, NY.: Springer; 2006. The Link between Inflammation and Cancer.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous