

The auxiliary subunit gamma 1 of the skeletal muscle L-type Ca2+ channel is an endogenous Ca2+ antagonist
- PMID: 17978188
- PMCID: PMC2077065
- DOI: 10.1073/pnas.0704340104
The auxiliary subunit gamma 1 of the skeletal muscle L-type Ca2+ channel is an endogenous Ca2+ antagonist
Abstract
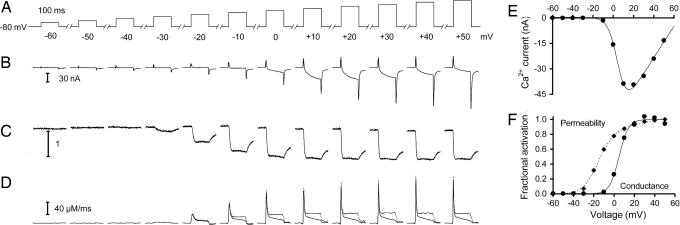
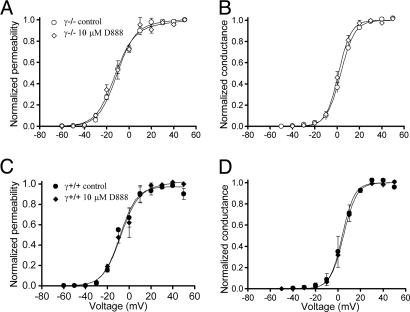
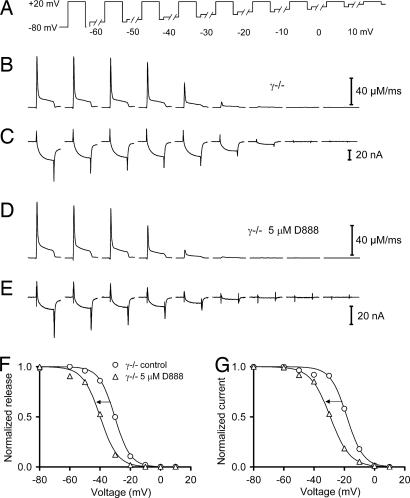
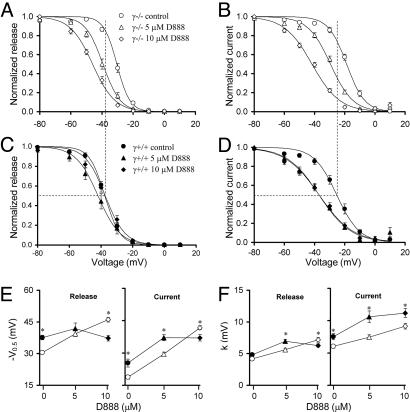
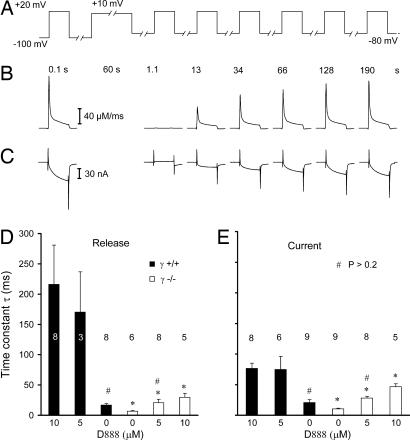
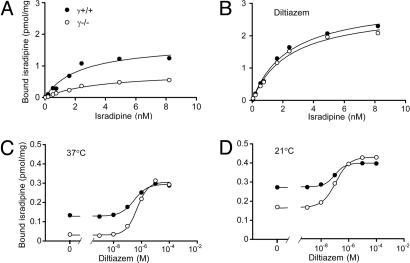
Ca2+ channels play crucial roles in cellular signal transduction and are important targets of pharmacological agents. They are also associated with auxiliary subunits exhibiting functions that are still incompletely resolved. Skeletal muscle L-type Ca2+ channels (dihydropyridine receptors, DHPRs) are specialized for the remote voltage control of type 1 ryanodine receptors (RyR1) to release stored Ca2+. The skeletal muscle-specific gamma subunit of the DHPR (gamma 1) down-modulates availability by altering its steady state voltage dependence. The effect resembles the action of certain Ca2+ antagonistic drugs that are thought to stabilize inactivated states of the DHPR. In the present study we investigated the cross influence of gamma 1 and Ca2+ antagonists by using wild-type (gamma+/+) and gamma 1 knockout (gamma-/-) mice. We studied voltage-dependent gating of both L-type Ca2+ current and Ca2+ release and the allosteric modulation of drug binding. We found that 10 microM diltiazem, a benzothiazepine drug, more than compensated for the reduction in high-affinity binding of the dihydropyridine agent isradipine caused by gamma 1 elimination; 5 muM devapamil [(-)D888], a phenylalkylamine Ca2+ antagonist, approximately reversed the right-shifted voltage dependence of availability and the accelerated recovery kinetics of Ca2+ current and Ca2+ release. Moreover, the presence of gamma 1 altered the effect of D888 on availability and strongly enhanced its impact on recovery kinetics demonstrating that gamma 1 and the drug do not act independently of each other. We propose that the gamma 1 subunit of the DHPR functions as an endogenous Ca2+ antagonist whose task may be to minimize Ca2+ entry and Ca2+ release under stress-induced conditions favoring plasmalemma depolarization.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
References
-
- Flaim SF, Zelis R. Calcium Blockers: Mechanisms of Action and Clinical Applications. Baltimore: Urban & Schwarzenberg; 1982.
-
- Fleckenstein A. Calcium Antagonism in Heart and Smooth Muscle. New York: Wiley; 1983.
-
- Melzer W, Herrmann-Frank A, Lüttgau HC. Biochim Biophys Acta. 1995;1241:59–116. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
