

Specificity in molecular design: a physical framework for probing the determinants of binding specificity and promiscuity in a biological environment
- PMID: 17979267
- PMCID: PMC3465720
- DOI: 10.1021/jp074285e
Specificity in molecular design: a physical framework for probing the determinants of binding specificity and promiscuity in a biological environment
Abstract
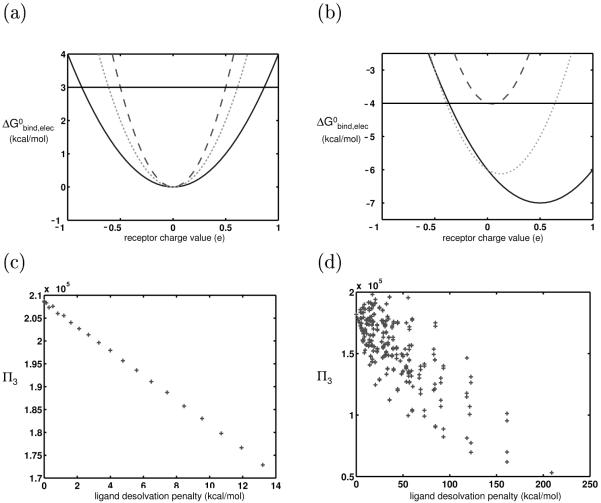
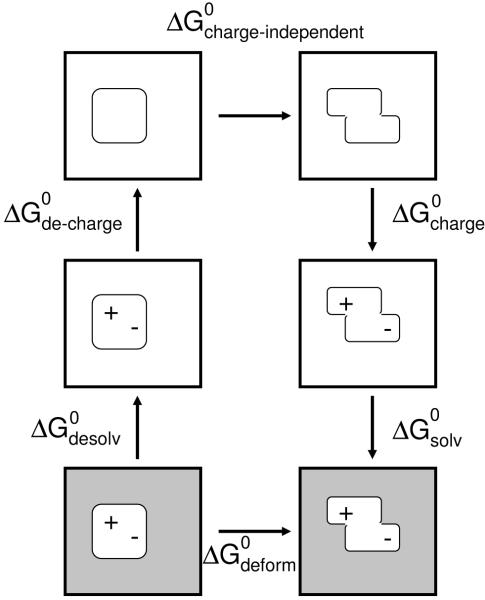
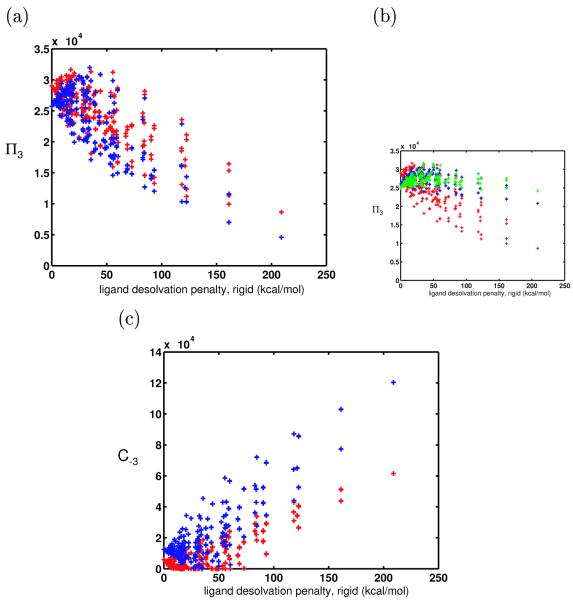
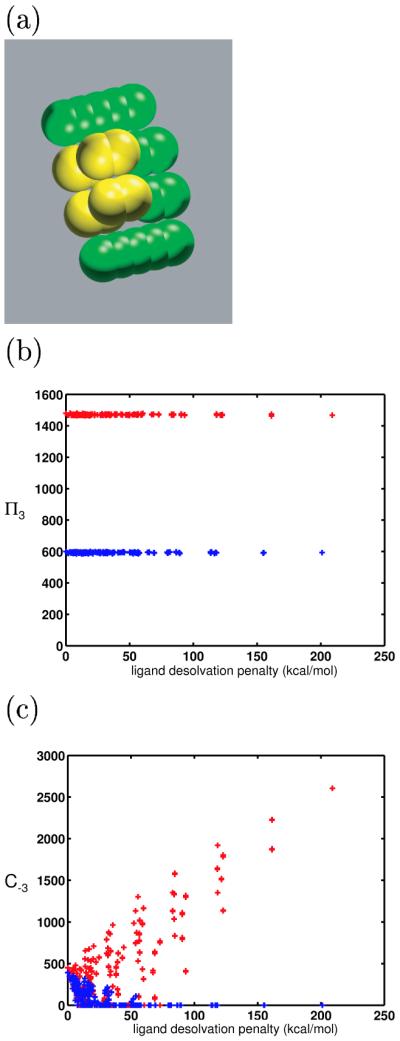
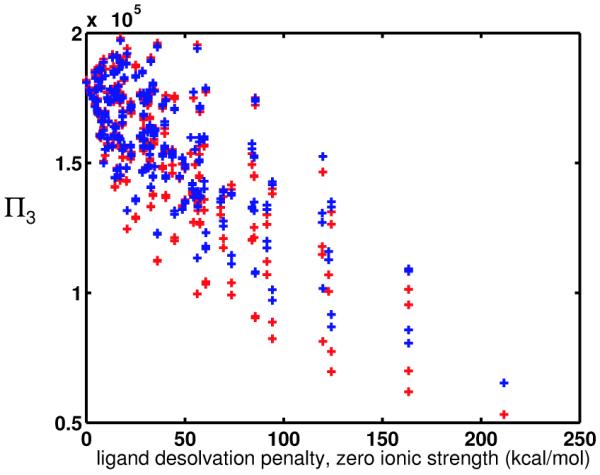
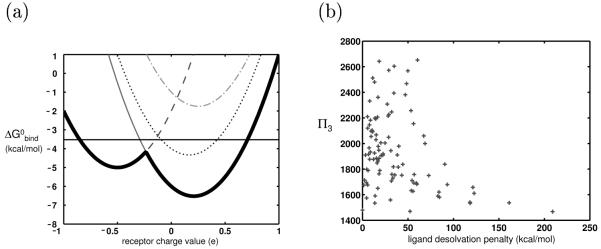
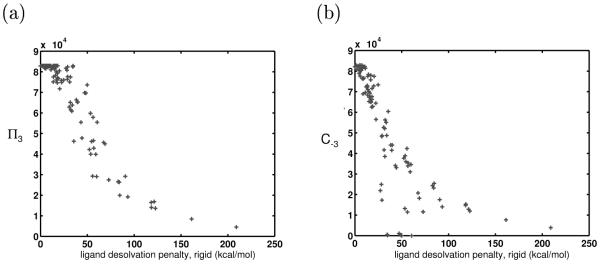
Binding specificity is an important consideration in drug design. An effective drug molecule often must bind with high specificity to its intended target in the body; lower specificity implies the possibility of significant binding to unintended partners, which could instigate deleterious side effects. However, if the target is a rapidly mutating agent, a drug that is too specific will quickly lose its efficacy by not binding well to functional mutants. Therefore, in molecular design, it is crucial to tailor the binding specificity of a drug to the problem at hand. In practice, specificity is often studied on a case-by-case basis, and it is difficult to create general understanding of the determinants of specificity from the union of such available cases. In this work, we undertook a comprehensive, general study of molecular binding with emphasis on understanding the determinants of specificity from a physical standpoint. By extending a theoretical framework grounded in continuum electrostatics and creating an abstracted lattice model that captures key physical aspects of binding interactions, we systematically explored the relationship between a molecule's physical characteristics and its binding specificity toward potential partners. The theory and simulated binding interactions suggested that charged molecules are more specific binders than their hydrophobic counterparts for several reasons. First, the biological spectrum of possible binding characteristics includes more partners that bind equally well to hydrophobic ligands than to charged ligands. Also, charged ligands, whose electrostatic potentials have strong orientational dependence, are more sensitive to shape complementarity than their hydrophobic counterparts. Ligand conformational and orientational flexibility can further influence a charged molecule's ability to bind specifically. Interestingly, we found that conformational flexibility can increase the specificity of polar and charged ligands, by allowing them to greatly lower the binding free energy to a select few partners relative to others. Additionally, factors such as a molecule's size and the ionic strength of the solution were found to predictably affect binding specificity. Taken together, these results, all of which stem from a unified theoretical framework, provide valuable physical insight into the general determinants of binding specificity and promiscuity in a biological environment. The general principles discussed here could prove useful in the design of molecules with tailored specificities, leading to more effective therapeutics.
Figures
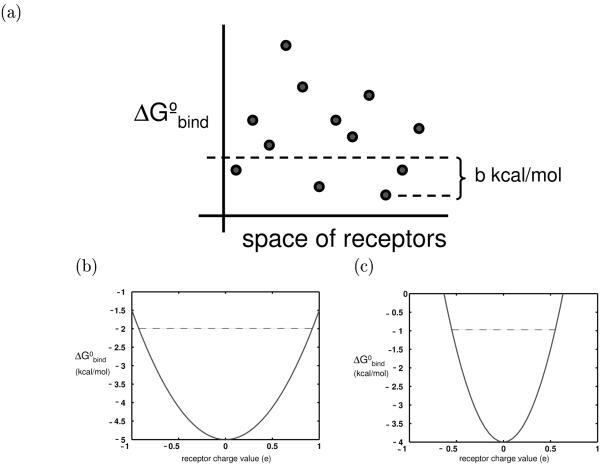

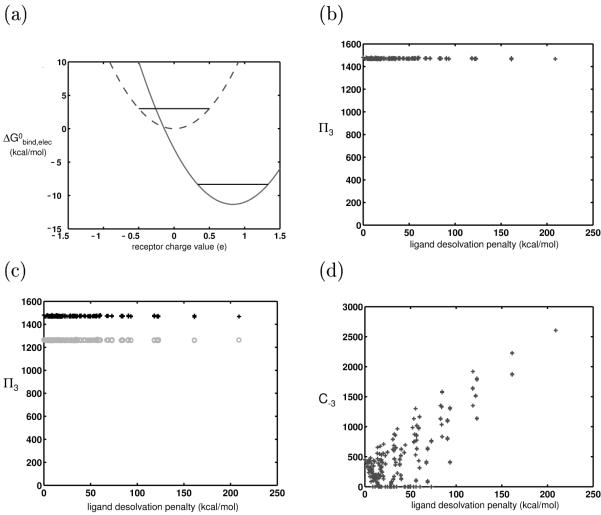
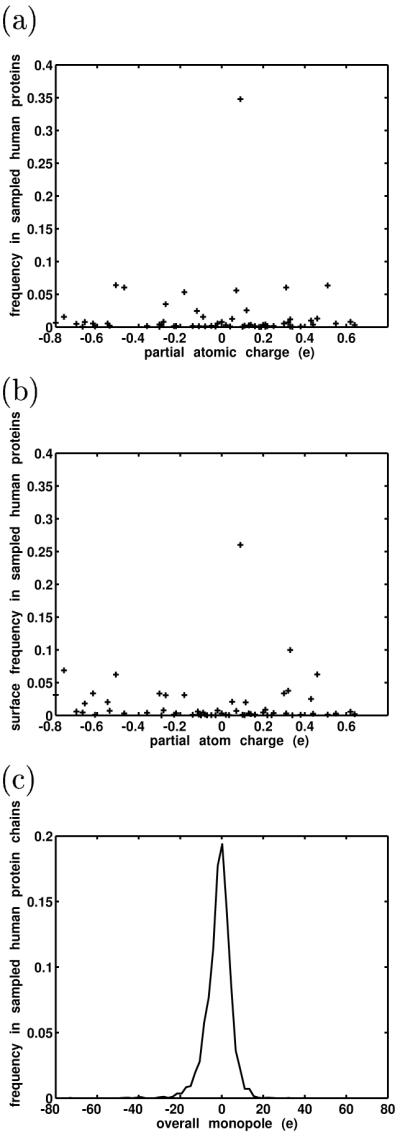
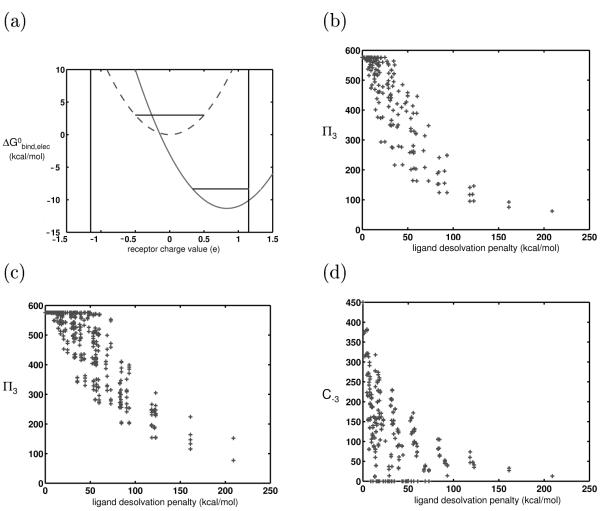
Similar articles
-
Macromolecular crowding: chemistry and physics meet biology (Ascona, Switzerland, 10-14 June 2012).Phys Biol. 2013 Aug;10(4):040301. doi: 10.1088/1478-3975/10/4/040301. Epub 2013 Aug 2. Phys Biol. 2013. PMID: 23912807
-
Electrostatic steering and ionic tethering in enzyme-ligand binding: insights from simulations.Proc Natl Acad Sci U S A. 1998 May 26;95(11):5942-9. doi: 10.1073/pnas.95.11.5942. Proc Natl Acad Sci U S A. 1998. PMID: 9600896 Free PMC article.
-
Entropic cost of protein-ligand binding and its dependence on the entropy in solution.J Phys Chem B. 2009 Apr 30;113(17):5871-84. doi: 10.1021/jp809968p. J Phys Chem B. 2009. PMID: 19351118
-
Ligand-protein docking: cancer research at the interface between biology and chemistry.Curr Med Chem. 2003 May;10(9):763-7. doi: 10.2174/0929867033457809. Curr Med Chem. 2003. PMID: 12678780 Review.
-
Electrostatics in protein binding and function.Curr Protein Pept Sci. 2002 Dec;3(6):601-14. doi: 10.2174/1389203023380431. Curr Protein Pept Sci. 2002. PMID: 12470214 Review.
Cited by
-
Rational approaches to improving selectivity in drug design.J Med Chem. 2012 Feb 23;55(4):1424-44. doi: 10.1021/jm2010332. Epub 2012 Jan 12. J Med Chem. 2012. PMID: 22239221 Free PMC article. Review. No abstract available.
-
Targeting ligand-gated ion channels in neurology and psychiatry: is pharmacological promiscuity an obstacle or an opportunity?BMC Pharmacol. 2010 Mar 2;10:3. doi: 10.1186/1471-2210-10-3. BMC Pharmacol. 2010. PMID: 20196850 Free PMC article.
-
Molecular mechanisms and design principles for promiscuous inhibitors to avoid drug resistance: lessons learned from HIV-1 protease inhibition.Proteins. 2015 Feb;83(2):351-72. doi: 10.1002/prot.24730. Proteins. 2015. PMID: 25410041 Free PMC article.
-
How to Model for a Living: The CSGF as a Catalyst for Supermodels.Comput Sci Eng. 2021 Nov-Dec;23(6):34-41. doi: 10.1109/mcse.2021.3119764. Epub 2021 Oct 14. Comput Sci Eng. 2021. PMID: 35600324 Free PMC article.
-
A Perspective on Interdicting in Protein Misfolding for Therapeutic Drug Design: Modulating the Formation of Nonlocal Contacts in α-Synuclein as a Strategy against Parkinson's Disease.J Phys Chem B. 2024 Jul 11;128(27):6439-6448. doi: 10.1021/acs.jpcb.3c07519. Epub 2024 Jun 28. J Phys Chem B. 2024. PMID: 38940731 Free PMC article. Review.
References
-
- Scapin G. Curr Drug Targets. 2006;7:1443–1454. - PubMed
-
- Freire E. Nat Biotechnol. 2002;20:15–16. - PubMed
-
- Voet D, Voet JG. Biochemistry. 2nd Ed. Wiley; New York: 1995.
-
- Lee JW, Beebe K, Nangle LA, Jang J, Longo-Guess CM, Cool SA, Davisson MT, Sundberg JP, Schimmel P, Ackerman SL. Nature. 2006;443:50–55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
